



图 1 是部分进入临床阶段的两个典型 PROTAC 实例,一个是招募 CRBN 口服给药的 ARV-110,一个是招募 VHL 静脉注射的 DT2216。
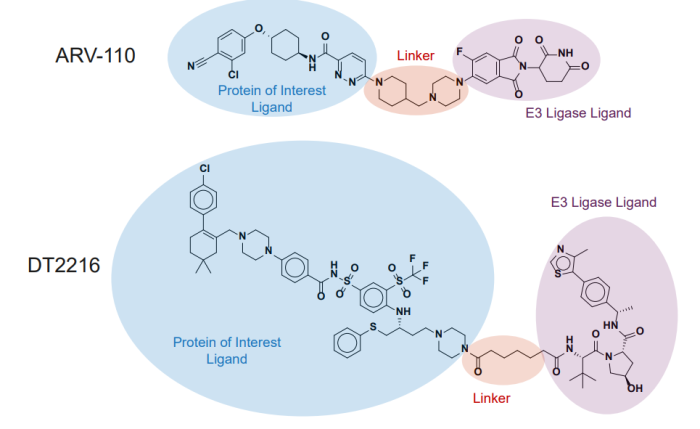
图 1
由于 PROTAC 构建方式的特殊性,其特征已经远远超过了常规口服吸收规则,IQ DMPK/ADME 工作组对 Janssen、Merck、Amgen、Novartis、AbbVie、AstraZeneca、Roche、GSK、Genentech 和 Takeda 等18 家制药企业进行调查。
该调查报告旨在通过汇集行业经验为 PROTAC 设计提供更有价值的信息,更快地将 PROTAC 推向临床,惠及患者。
行业报告由以下 64 个调查问题的答案整理而来,所有回答仅代表各企业调查人的个人观点。
调查问题(向下滑动查看更多)
1. in your experience, do targeted protein degraders obey the free drug hypothesis at a practical time scale with a normal dosing regimen?
2. When there is a disconnect from the free drug hypothesis, have in vitro assay lC50/EC50 values also been corrected for protein binding?
3. Has an explanation been identified for the disconnect?
4. Are you developing targeted protein degraders for the oral route of administration?
5. What formulation strategies have you used most often and/or have been most successful for initial in vivo PK studies using the oral route of administration for targeted protein degraders at each stage of development? Select all that apply.
6. In your experience, what polar surface area (PSA) is most likely to result in an orally bioavailable targeted protein degrader?
7. In your experience, what LogD (pH 7.4) is most likely to result in an orally bioavailable (>30% F) targeted protein degrader?
8. In your experience, what molecular weight is most likely to result in an orally bioavallable targeted protein degrader?
9. ln your experience, what solubility (uM) at pH 7.4 is most likely to result in an orally bioavailable targeted protein degrader?
10. In your experience, how many H-bond acceptors are most likely to result in a successful oral targeted
protein degrader?
11. In your experience, how many H-bond donors are most likely to result in a successful oral targeted
protein degrader?
12. ln your experience, how many rotatable bonds are most likely to result in an orally bioavailable targeted protein degrader?
13. What other properties do you consider when developing an orally bioavailable (>30% F) targeted protein
degrader?
14. What are the top 1 or 2 ADME/physical property challenges you encounter when developing a targeted
protein degrader with good oral PK (>30% F)?
15. Would your property range answers differ for a successful intravenous targeted protein degrader?
16. How would property ranges differ for intravenous compared with oral degraders?
17. ls your ADME assessment of targeted protein degraders different from standard small molecules with poor
physico-chemical properties (i.e., that break the Rule of 5)?
18. What are the most significant changes you have made to the ADM assessment assays?
19. Please briefly describe the general principles of new assays that you have successfully used.
20 have you modified your DDI assessment due to the potential degradation of enzymes/transporters?
21. Are there any unique protein degrader properties that influence your species selection for in vivo ADME and PK studies?
22. Are your strategies to predict human PK for targeted protein degraders different than traditional small molecules?
23. what strategics do you use for huam PK prediction of target protein degrader?
24. In your experience, do standard preclinical PK prediction methods (e.g., in vitro to in vivo extrapolation[IVIVE]) work as well for targeted protein degraders as for typical small molecules?
25. If no, what is the suspected or known cause of discrepancies?
26. Have you assessed the potential for lymphatic absorption of targeted protein degraders?
27. Have you used intravenous dosing for targeted protein degraders?
28. what issues were observed with IV dosing?
29. Have you used subcutaneous dosing for targeted protein degraders?
30. what issues were observed with SC dosing?
31. ls there any unique pharmacological impact of the metabolites of targeted protein degraders?
32 what unique observations were noted?
33. What major clearance mechanism(s) have you observed with targeted protein degraders? Select all that apply.
34. Do you have experience with targeted protein degraders that cross the blood-brain barrier?
35. What is the range of observed unbound Kp (brain:plasma) values of these BBB crossingdegraders?
36. Have you observed any limitations to distribution besides the BBB for targeted protein degraders?
37. Please describe other tissues in which you have observedlimitations.
38. What method(s) do you use most often to measure target protein resynthesis?
39. Have you modified your in vitro ADME assays (e.g, solubility, plasma protein binding, permeability, intrinsicclearance, etc.) to improve data quality (e.g., accuracy, reproducibility, etc.) for targeted protein degraders?
40. Briefly describe the general modifications that have been made to assays (ie, no need to provide % modifiervalues or specific times).
41. In your experience, what plasma protein binding range(s) do targeted protein degraders fall within? Selectall that apply
42. Please rank the plasma protein binding ranges from most common bound to least common bound that mosttargeted protein degraders fall within
43. Are your methods for measuring plasma protein binding for targeted protein degraders different thantraditional small molecules?
44. What method(s) do you use most often to measure plasma protein binding for targeted protein degraders?
45. What is/are your top challenge(s) to accurately measuring plasma protein binding for targeted proteindegraders?
46. Have you experienced epimerization/racemization with targeted protein degraders?
47. Did vou handle epimerization/racemization of targeted protein decraders differently than vou would have foia standard small molecule?
48. What did you do differently for targeted proteindegraders?
49. In your experience, do the properties of a targeted protein degrader's individual structural parts (i.e., targetprotein ligand, linker, or E3 ligase ligand) dictate the overall properties (e.g., permeability, intrinsic clearanceetc.) of the whole (intact) degrader (i.e., have you established a correlation between parts and properties)?
50. What properties have you optimized on the parts?
51. Do you have examples of parts having poor properties, but the whole (intact) degrader having improvedproperties and/or oral PK?
52. Did optimization of the linker cause the improved properties for the whole (intact) degrader?
53. What ranges) of target protein resynthesis rates have you most often observed for targeted proteindegrader targets?
54. Do you consider ADME concerns and/or drug-ability when selecting an E3 ligase?
55. If not ADME concerns and/or drug-ability, what quality do you use to drive ligases election?
56. Approximately how many in vivo efficacy studies (e.g., PK/PD, tumor growth inhibition, etc,) have you run with targeted protein degraders?
57. How many examples of the hook effect have you observed during in vivo efficacy studies (e.g., PK/PDtumor growth inhibition, etc.)?
58. Were unbound plasma concentrations measured to confirm the hook effect during in vivo eficacy studies?
59. Were unbound concentrations in the target tissue measured to confirm the hook effect during in vivoefficacy studies?
60. What interpretations were impacted by the observation of the hook effect in vivo? Select all that apply
61. How much experience have you had with in silico/machine learning prediction of targeted protein degrader ADME properties?
62. In your extensive experience, have in silico tools accurately predicted the ADME properties of targetedprotein degraders?
63. How much experience have you had with PBPK prediction of PK for targeted protein degraders?
64. In your extensive experience, have PBPK tools (e.g., SimCYP, GastroPlus, etc,) accurately predicted thePK of targeted protein degraders?
口服 TPD 的一般工作流程
18 家制药企业中有 17 家正在开发用于口服给药的降解剂,根据回复,实现降解剂口服生物利用度的最佳化学性质统计为:tPSA=100-200 Å2、MW <900 Da、氢键受体 (HBA)<14、氢键供体 (HBD) <5 和 LogD=3- 5(图 2A-G)。溶解度和可旋转键的数量在调查中分布更为均匀。
18 家回复公司中的 15 家采用了静脉注射的给药途径。其中,七个报告的问题主要与由于 PROTAC 在所需剂量水平和给药体积下的溶解度差而导致的制剂挑战有关。大多数公司报告说,与口服相比,IV 给药途径探索了相似或更广泛的理化特性(图 2H)。logD和溶解度是两个例外。
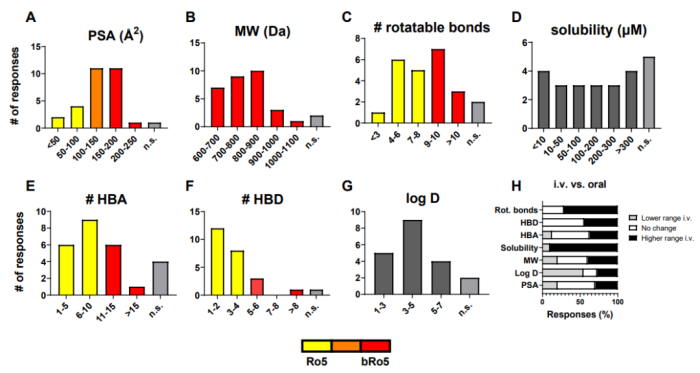
图 2 口服和静脉注射TPDs的最佳性能范围
据回复,渗透性、溶解度和 MW 是开发口服 PROTAC 时要克服的主要挑战。次要挑战包括代谢不稳定性、氢键供体数量、极性/亲脂性以及最大限度地减少 P-gp 对吸收的影响(图 3)。
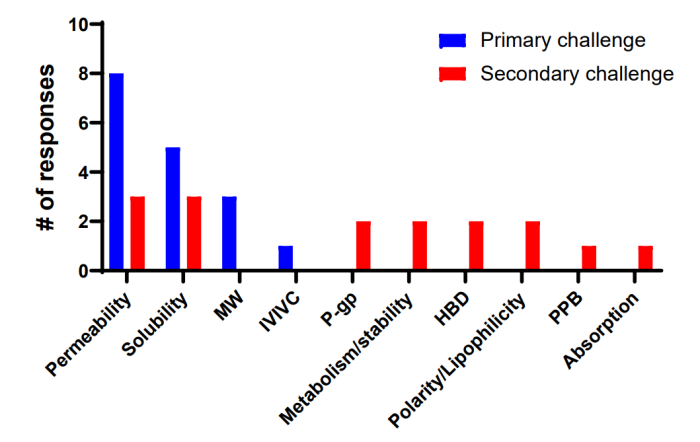
图 3 口服给药面临的ADME/物理化学性质挑战
溶解度是受访者列出的一个关键参数,用于改善静脉和口服给药途径的暴露和耐受性。对于静脉PROTAC 使是那些具有高渗透性的小分子,低水溶性也会限制小分子的口服生物利用度。
尽管如此,约 40% 的 FDA 批准的口服药物属于生物药剂学分类系统(BCS)类别中的低溶解度类BCS IV 类约占批准药物的 7%)。

如前所述,当渗透性或溶解度较低时,P-gp 的主动外排会进一步限制口服生物利用度
18 家回复公司中有 11 家使用了皮下注射 SC 的给药途径,这可能在临床前使用以实现早期化合物的充分暴露。在那些皮下给药 PROTAC 报告的问题包括受试者间的高变异性、高剂量下暴露线性缺失、配方挑战、水肿形成、坏死、休克样反应、局部炎症和缺乏跨物种的可转译性。
——降解剂 ADME 评估过程中工作流程的修改——
Absorption:
口服给药肯定是最优选的给药途径,对接受长期治疗的患者很方便,对医生调整给药方案也方便。但是 PROTAC 这种高分子量的化合物实现口服给药就面临着重大挑战,但临床上有许多高分子量的口服 PROTAC。
据受访者提供的信息表明,一种可能的口服 PROTAC 吸收替代机制是通过淋巴吸收。一些生物制药分类系统 (BCS) IV 类和一些 bRo5 化合物(例如维奈托克)具有适度的口服生物利用度,部分原因就是淋巴吸收。
当一种化合物与胃肠道中乳糜微粒的脂质/甘油三酯结合,并与肠细胞中的乳糜微粒一起被加工以分泌到淋巴循环中,从而绕过首过代谢时,就会发生口服化合物的淋巴吸收。通常,淋巴吸收的化合物具有高 HBA、PSA、Log P 和 MW。
18 家公司中有 4 家评估了降解剂的淋巴吸收潜力。其中,只有 2 个观察到淋巴吸收。在这些淋巴吸收研究中,没有完成随访以确定哪种给药途径与哪种结果相关,但根据作者的经验,可以通过口服、舌下或皮下给药观察到淋巴吸收。需要进一步研究以确定口服 PROTAC 是否可以被淋巴吸收,以及是否可以利用这种机制来增加口服生物利用度。
Tissue distribution :
18 家公司回答了有关 PROTAC 组织分布的问题(除了脑)。8 家公司表示他们没有投入资源来测量组织浓度,而从事组织分布分析的 10 家公司中有 9 家表示组织浓度接近或高于预期。其余受访者表示肿瘤水平低于预期,并表明这可能已通过 P-gp 介导的外排限制分布到肿瘤细胞。
Plasma protein binding:
血浆蛋白结合 (PPB) 是一种药物代谢和药代动力学 (DMPK) 参数,对于解释药物特性(例如清除率和分布体积)以及开发药物-药物相互作用预测和药代动力学-药效学(PKPD) 关系至关重要。已经开发了许多技术来测量药物血浆蛋白结合和游离药物浓度。传统小分子最常用的技术是平衡透析、超速离心和超滤。
药物分子的挑战,例如高非特异性结合(即与孵育板或试管塑料的结合)、血浆不稳定或达到平衡所需的长时间孵育对于小分子来说很常见。重要的是要考虑每种 PPB 技术的各种优点和注意事项,以及可能需要改进结果的各种检测调整。
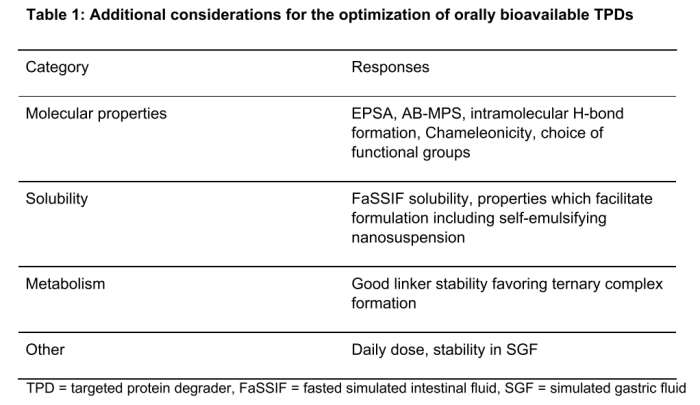
总共有 18 家公司回答了与 PROTAC PROTAC 小于 50% 到大于 99.9%。血浆蛋白结合的最高发生率在 99 - 99.9% 范围内(图 5)。
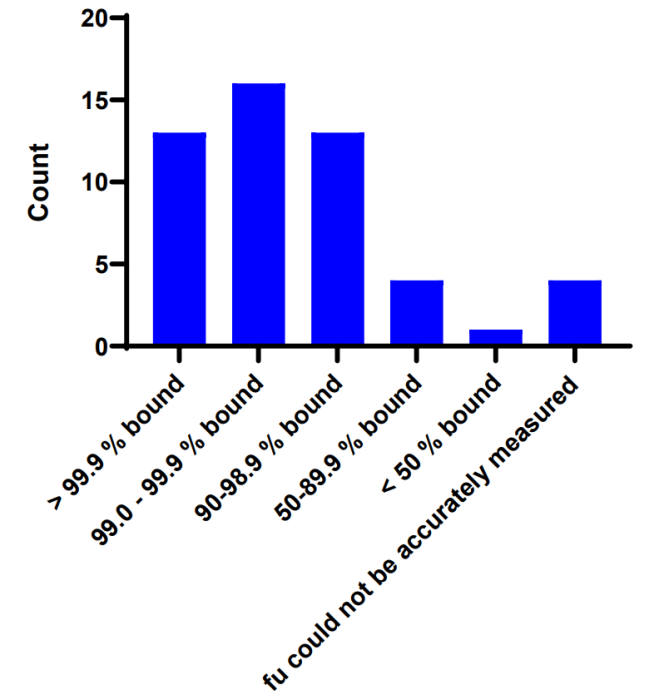
图 5 目标蛋白降解剂 PPB 观察范围
The free drug principle:
Free Drug Principle 是一个成熟的小分子概念,在解释 PK/PD 关系时广泛应用于药物发现和开发。调查回复表明,大多数公司都认为治疗靶点的稳态游离浓度与 PROTAC 的药效学效应之间存在明确的关系。
17 名受访者中有 3 名表示虽然没有主要的 PK/PD 脱钩,但观察到轻微的脱钩,这可能是由于 TPD 的膜渗透率低,导致在目标细胞的质膜上达到平衡的时间更长。这或许可以通过在体外测定中测量未结合的细胞效力来解决。因为药物的未结合浓度不仅决定了药物的药理作用 (PD),还决定了其在药物中的总清除率和分布,所以需要准确测定 PPB 才能充分了解传统小分子以及 bRo5 化合物(如 PROTAC)的 PK和 PK/PD 关系。
重要的是要注意,PROTAC PROTAC 的关键特征之一,可以平衡分子的一些 ADME 挑战。
Target half-life:
14 名测量目标蛋白再合成率的受访者称,观察到最多的范围是 9-24 小时 (9/14),其次是 大于 24 小时 (5/14)。多名受访者表示,他们很少研究再合成率小于 2 小时和 2-8 小时范围内的目标。
Hook effect:
当与 POI 或 E3 配体结合饱和时,在高体外浓度的 PROTAC 下观察到 Hook 效应,导致形成无活性的二元复合物,从而导致钟形 DC50 曲线。受访者使用 TPD 进行了超过 350 项体内 PK/PD 或疗效研究。在这些研究中,只有 2 个受访者在体内观察到了 6 个 Hook 效应的例子。然而,在这些研究中未直接测量未结合的血浆或组织浓度以确认数据可以用钩状效应解释。这些受访者表示,假定的 Hook 效应确实对数据解释、PK/PD 建模和人体有效剂量预测有影响。
Brain penetration:
血脑屏障由具有非常紧密的细胞间连接的内皮细胞组成,这限制了药物的细胞旁通量。药物穿过血脑屏障的通道高度依赖于药物的理化特性、被动扩散以及摄取和外排转运蛋白。由于 TPD 的物理化学特性,实现药理学相关的大脑或中枢神经系统 TPD 浓度是一项重大挑战。
17 家公司回答了与 PROTAC PROTAC PROTAC PROTAC 的脑渗透潜力较低。对于测量生物学相关脑浓度的 3 家公司,相应的未结合脑与血浆 (B/P) 比率分别为 0.1-1.0、0.1-2.0 和 0.1-35。
作者匿名从提供脑/血浆比为35的受访者那里收到了额外信息,对于该化合物,受访者确认该比率是未结合的脑血浆比 Kpuu,brain(不是总 Kp),这个结果在多项后续研究中得到证实,平均 Kpuu,brain 为 17。此外,受访者指出,他们所有其他 Kpuu,brain 测量值都在 0.1 到 1 的范围内。
图 6 TPDs 脑浓度评估。BLQ即低于定量限度
低 Kpuu,brain 0.1 值不足为奇,可能是由于血脑屏障的活跃流出。Kurimchak 等人最近表明 P-gp siRNA 和抑制剂可以降低由 Mdr1 (P-gp) 过表达介导的肿瘤细胞系耐药性。尽管他们没有直接证明 P-gp 介导的化合物流出,但这份报告连同调查答复表明 PROTAC 可以是 P-gp 底物。Kpuu,brain 为 1 支持开发 CNS 渗透性降解剂。Kpuu,brain 为 35 表明转运蛋白可能会有主动摄取。
据报道,bRo5 化合物 bromocriptine(MW 655 Da)是 OATP2B1 和 OATP1A2 的底物,表明这些转运蛋白可以转运高分子量化合物。其中一位受访者指出,他们观察到 OATP 转运蛋白主动摄取 TPD 作为主要清除机制,因此 OATP 主动摄取似乎是可行的。虽然这一领域需要做更多的工作,但口服 CNS 活性 PROTAC 似乎有希望。
Metabolism and elimination:
18 人中有 17 人观察到细胞色素 P450 (CYP450) 介导的代谢。18 名受访者中有 7 名将 UGT 催化的葡萄糖醛酸化视为主要代谢途径。CYP450 和 UGT 酶也是传统小分子的常见代谢途径。
8 名受访者分别选择酰胺酶和酯酶活性作为主要代谢酶,而 18 名受访者中的 3 名选择水解作为主要消除途径。由于 PROTAC PROTAC PROTAC 清除的酶,2 人提到了醛氧化酶,1 名受访者提到了谷胱甘肽 S-转移酶 (GST)(图 7)。
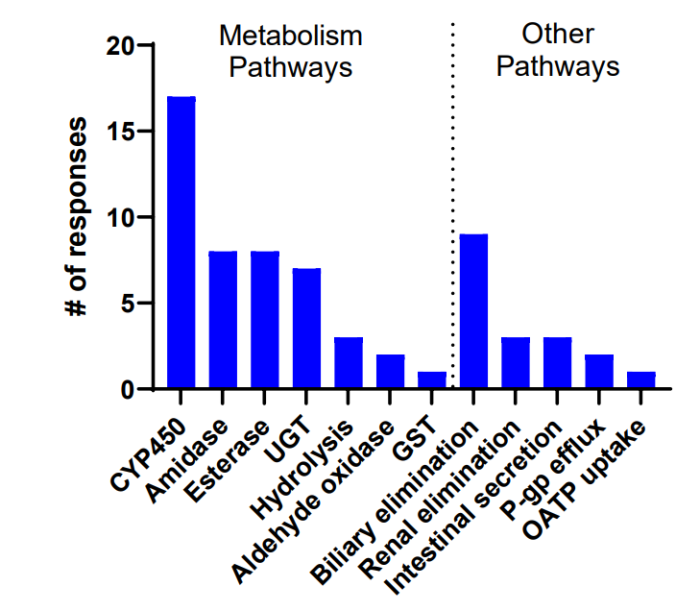
图 7 受访者观察到的主要消除机制
关于 PROTAC 的非代谢消除的文献仅限于一份报告,该报告是对大鼠施用高清除率分子,约 9% 的 IV 剂量作为母体排泄到胆汁中,排泄到粪便或尿液中的量可忽略不计。然而,一半的受访者说他们观察到胆汁排泄,18 人中的 3 人观察到肾脏排泄。在一份报告中,由于 PROTAC PROTAC 不太可能通过肾脏消除,但根据调查答复,肾脏排泄是可能的。
18 名受访者中的 3 名选择转运蛋白介导的消除作为观察到的消除机制,3 名中的 2 名指的是P-gp 外排,1 名指的是有机阴离子转运多肽 (OATP) 介导的肝摄取 . 18 名应答者中有 3 名在未提及特定转运蛋白的情况下将肠道分泌物列为主要的消除途径。在肠道中,几种转运蛋白可能参与肠道的主动分泌,但结合胆汁排泄的高观察率和已知的 PROTAC P-gp 外排,P-gp 很可能参与,但不能确定 MRP2 和 BCRP 的参与。
此外,受访者还被问及代谢物是否影响 PROTAC 的药理学PROTAC 进行 PK/PD 研究解释。
Modifications necessary:
89% 的受访者报告使用了修改后的体外 ADME 检测方法。令人惊讶的是,没有受访者报告对他们的药物相互作用评估策略进行了修改,但回复率仅为 44%。在标准 ADME 分析中降解剂遇到的典型挑战包括非特异性结合、低稳定性和缺乏溶解性(表 2)。
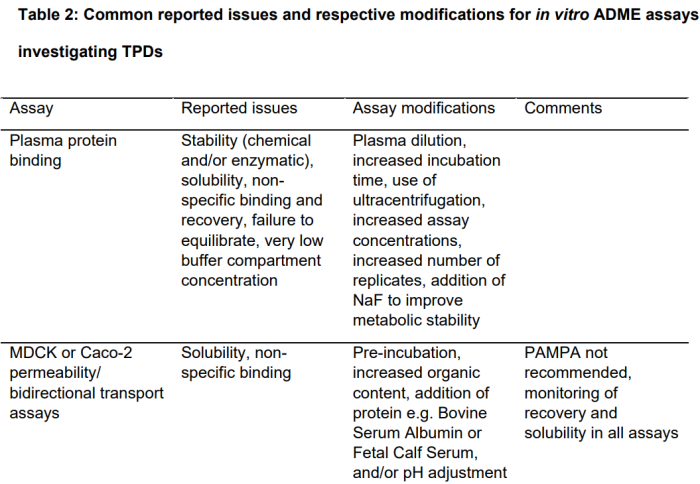
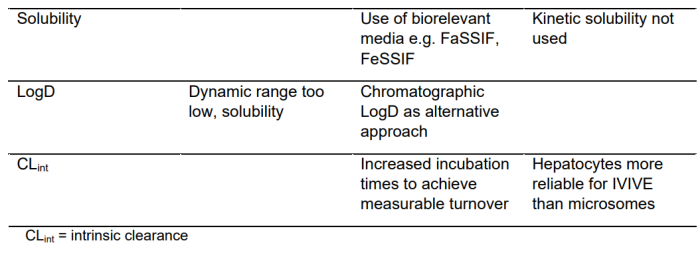
低溶解度会影响可在测定中测试的最大浓度,导致测试结果发生变化。与实验室器具的非特异性结合以及与孵育组分的蛋白质结合也可能导致游离浓度低于理论浓度,特别是在低化合物浓度下。
这两个因素导致了一个狭窄的浓度范围,在这个范围内进行体外实验没有混淆效应。添加牛血清白蛋白或溶剂等修饰剂可用于某些测定,例如渗透率或肝细胞 CLint,但必须注意不要干扰测定性能。除了这些修改,一些受访者还建立了额外的检测方法来评估降解剂的理化性质(如EPSA、分子内 H 键形成和极性基团遮蔽能力)或确定亲油性和渗透性。这样的分析可以避免通常描述的非特异性结合和溶解度问题。此外,AB-MPS 分数也被提到可以作为评估降解剂的生物利用度。一位受访者指出,不推荐平行人工膜通透性测定 (PAMPA),文献对 PAMPA 的运用也有矛盾的观点。
特别是对于 PPB,受访者采用了多种 PPB 方法来解决已知的检测挑战。受访者对 PPB 方法的首选是快速平衡透析(RED)或高通量透析(HT 透析),接下来是超速离心(25%)、传统平衡透析(15%) 和通量透析(5%),另一种很少使用的方法是超滤。在受访者中,55.6% 表示他们测量血浆蛋白与降解剂结合的方法不同于传统的小分子;然而,没有说明用于测量血浆蛋白结合的装置是否与用于传统小分子的装置不同。
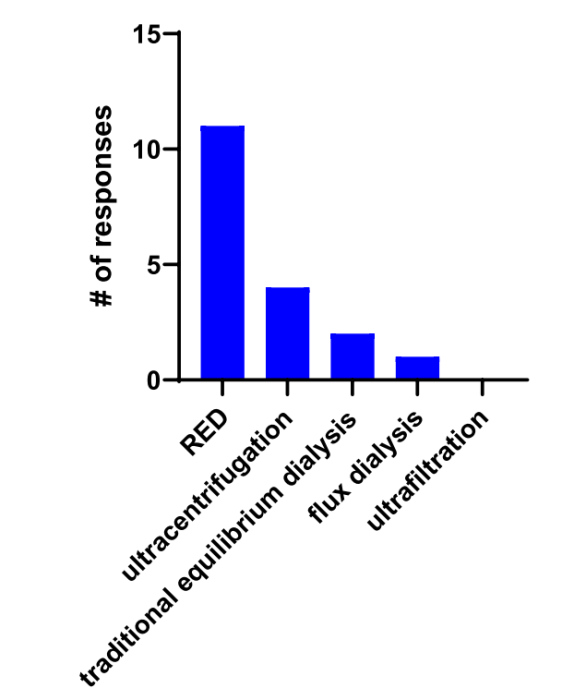
S2 测定 PPB 常用的方法
受访者提出测量 PROTAC 的 PPB 相关的主要挑战,如图 8 所示。其中最常见的是与塑料制品或透析膜的高度非特异性结合和相关的低的测定回收率 (45%),其次是分析灵敏度限制,特别是对于非常高的结合剂 (21%)、血浆不稳定 (14%),最后是未达到平衡 (7%) 或由于结合缓慢的结合动力学 (3%)。
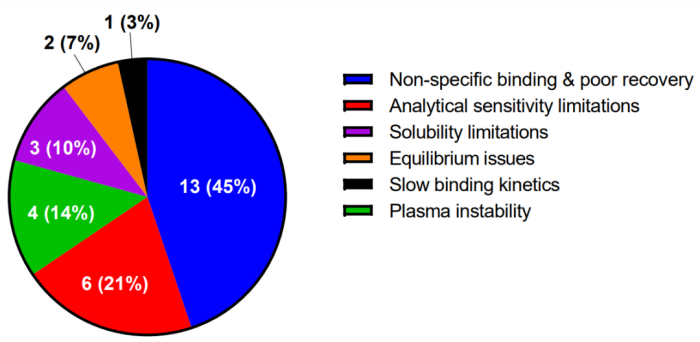
图8 测量 degrader PPB 面临的主要挑战
此外,受访者列出了在测量 PROTAC PROTAC 通常具有高于 99% 的 PPB)。
PROTAC 的化学和代谢稳定性也会混淆体外 ADME 测定结果的解释。具体而言,沙利度胺等 CRBN 配体已被证明在中性 pH 条件下会发生化学水解,并且戊二酰亚胺环也会发生依赖于物种的代谢水解。添加代谢抑制剂的 RED(如防止酰胺水解的氟化钠)、通量透析、超滤也提供了测量代谢不稳定的 PROTAC PPB 的选择。
这些测定修改最初是为了满足对非 TPD bRo5 化合物的需求而实施的。从制剂的角度来看,在各公司的初始PK研究中,通常采用一系列口服制剂策略,主要是在先导发现和先导优化期间。补充图S3中显示了这些策略的详细信息以及它们通常采用的阶段。根据作者的经验,这些配方策略与传统的小分子相当,特别是bRo5。
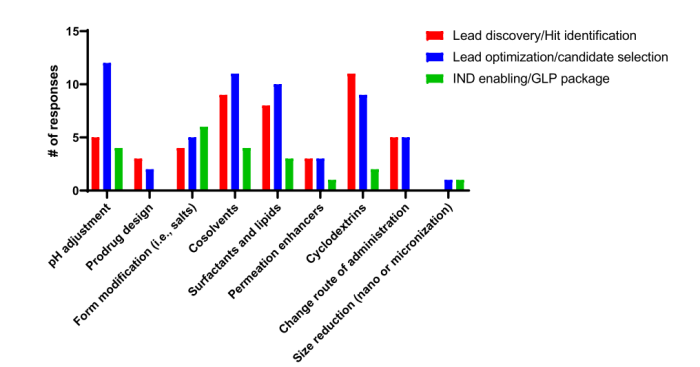
S3 初步PK研究中最成功的制剂策略
In vivo species selection
传统小分子毒理学研究的物种选择通常是通过比较临床前物种与人类的体外代谢谱、代谢物和药理学来完成的。大多数 (11/17) 响应者表示,PROTAC PROTAC 的物种选择与传统小分子没有根本区别。
optimize PK / ADME properties
受访者被问及他们是否检查了 PROTAC 各成分(即E3 配体、POI 配体、接头)对 PROTAC 整体 PK 特性的贡献。18 人中有 7 人表示他们没有尝试,有 3 人表示他们将单个成分的特性与 PROTAC 的特性进行了比较,但无法建立相关性,8 名检查了 PROTAC 各个组成部分的物理特性的作用,并能够建立相关性。
受访者还被问及他们是否观察到 TPD 相对于 TPD 内的各成分具有改进的 PK 特性。在 11 个回答中,有 7 个回答“否”,而 4 个回答“是”。这表明 TPD 的 PK 属性通常并不比其组件更好。
受访者被要求列出他们如何优化 TPD的组件,收到了 9 份答复。一位受访者简单地列出了 TPD 的三个组成部分(连接酶结合物、接头、目标结合物)。3 位回复者列出了 ADME/理化特性(渗透性、HBD、MW、代谢稳定性、溶解度),另外 1 位回复者将”转运蛋白特性“列为改进的重点。
三位受访者提到了 Linker 的特性,特别是 Linker 与其他组件的连接、Linker 长度和类型、Linker Linker Linker Linker 的改进可改进 PROTAC 整个性能。
此外,一位受访者将连接酶结合部分对水解的稳定性列为改进的重点。受访者被问及在选择 E3 连接酶时是否考虑了 ADME 特性或药物能力。在 18 个回答中,14 个表示是的,ADME 和成药性是选择 E3 连接酶的关键考虑因素。4 名受访者表示 ADME 不是选择 E3 连接酶的关键问题。
Prediction of PK
由于 PROTAC 刚刚进入临床,因此很少公开披露成功预测人体药代动力学,特别是人体清除率。17 人中的 14 人表示他们预测降解剂 PK 的方法与传统小分子所采用的方法没有区别。尽管 17 名受访者中有三名表示采用了替代策略,但没有指定传统策略的替代方法。
有趣的是,人类 PK 预测最常见的策略是 allometry,其次是 IVIVE,9 名受访者表示他们使用了 PBPK 建模(图 9 a)。
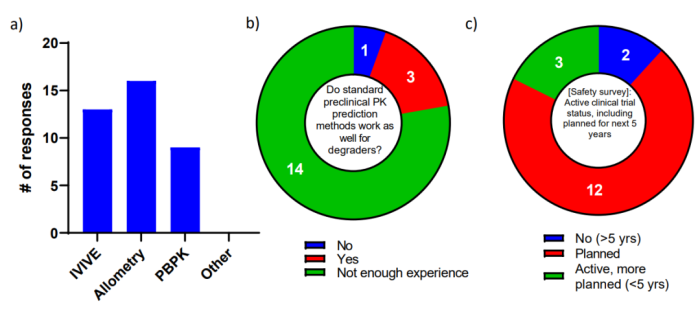
图9 PROTAC PK 预测
在将小分子的体外 CLint 缩放到体内 CL 时实现IVIVC 可提高预测人体清除率的信心。然而,当涉及非 P450 清除途径时,通常不会观察到 IVIVC 方法。在这种情况下,allometry 通常是替代方法,其次是 PBPK 建模或与 PBPK 建模相结合,这三种方法可以同时使用。
微粒体、肝细胞和血浆蛋白结合都很难测量,因为 PROTAC 尽管如此,已经报道了一系列使用小鼠肝细胞的 TPD 的良好 IVIVC(大多数在3 倍以内),这表明通过仔细测量,IVIVE 可以成为 TPD的有用/有效方法。
当被问及这些标准的临床前方法是否像它们对典型的小分子一样对蛋白质降解剂起作用时,18 名受访者中超过 75% 的人表示没有足够的经验来根据当前可用的数据进行比较(图 9 b) . 事实上,迄今为止,关于降解剂的临床药代动力学数据的披露非常有限。在回答平行调查中的一个问题时,只有三名受访者进行了积极的持续临床试验,其中大多数人计划在未来五年内进行临床研究(图 9 c)。随着更多临床数据的出现,这将成为后续调查的一个重要领域。
In silico tools and PBPK modeling:
18 名受访者中有 12 名在使用计算/机器学习预测降解剂的 ADME 特性方面有一些经验,其中一名受访者拥有丰富的经验,其经验表明他们有时可以准确预测 TPD 的 ADME 特性。相比之下,使用 PBPK 进行 PK 预测似乎有限,17 名受访者中有 11 名没有经验,大约三分之一有一些经验,一名受访者有丰富的经验。一位经验丰富的受访者报告说,PBPK 方法可以准确预测 TPD 的 PK。
调查答复中有两个明显的主题:1) 口服递送是首选并且可以通过 TPD 实现,2) TPD 的DMPK/ADME 评估与其他 bRo5 化合物没有显著差异。
内容来源:FenDiPharma 责任编辑:胡静 审核人:何发
声明:本文内容由平台创作者发布,内容仅代表作者本人观点;如内容涉及违法、侵权等情形,请及时联系工作人员处理!工作人员微信:pckt6842。

