

达比加群酯( dabigatran etexilate) 是作用于Ⅱa因子靶点的新一代口服直接凝血酶抑制剂,可预防血栓形成,其活性基团还可抑制游离凝血酶、与血块结合的凝血酶和凝血酶诱导的血小板聚集]。达比加群酯口服后经胃肠道吸收,给药后0. 5 至2. 0 h达到峰浓度( cmax) 。达比加群酯胶囊已在多个国家上市,具有无需常规监测凝血、并发症少、药物相互作用少等优点,临床使用日益广泛,成为目前国内抗凝血药物仿制药开发领域的热点。
达比加群酯胶囊的活性成分为甲磺酸达比加群酯,结构式β-丙氨酸( 丙氨酸),N-[[2-[[[4-[[[( 己氧(氧)基) 羰基]氨基]亚氨基甲基]苯基]氨基]甲基]-1-甲基-1H-苯并咪唑-5-基]羰基]-N-2-嘧啶-,乙酯,甲磺酸盐( 图1) ,其在水中的溶解度为1. 8 mg·mL - 1,易溶于甲醇,可溶于乙醇(乙醇),微溶于异丙醇(异丙醇)及丙酮,几乎不溶于乙酸乙酯(乙酸乙酯)。达比加群酯溶解性具有pH 依赖性,pH 低于3 时溶解性较好,体内吸收也会受到制剂微环境的影响,同时,达比加群酯结构并不稳定,酸性、碱性环境中可能发生因酯键水解导致的活性成分降解。因此制剂的处方工艺设计及生产过程控制,可能对制剂的关键质量属性以及体内作用的发挥起到重要的作。
丙氨酸),N-[[2-[[[4-[[[( 己氧(氧)基) 羰基]氨基]亚氨基甲基]苯基]氨基]甲基]-1-甲基-1H-苯并咪唑-5-基]羰基]-N-2-嘧啶-,乙酯,甲磺酸盐( 图1) ,其在水中的溶解度为1. 8 mg·mL - 1,易溶于甲醇,可溶于乙醇(乙醇),微溶于异丙醇(异丙醇)及丙酮,几乎不溶于乙酸乙酯(乙酸乙酯)。达比加群酯溶解性具有pH 依赖性,pH 低于3 时溶解性较好,体内吸收也会受到制剂微环境的影响,同时,达比加群酯结构并不稳定,酸性、碱性环境中可能发生因酯键水解导致的活性成分降解。因此制剂的处方工艺设计及生产过程控制,可能对制剂的关键质量属性以及体内作用的发挥起到重要的作。
丙氨酸),N-[[2-[[[4-[[[( 己氧(氧)基) 羰基]氨基]亚氨基甲基]苯基]氨基]甲基]-1-甲基-1H-苯并咪唑-5-基]羰基]-N-2-嘧啶-,乙酯,甲磺酸盐( 图1) ,其在水中的溶解度为1. 8 mg·mL - 1,易溶于甲醇,可溶于乙醇(乙醇),微溶于异丙醇(异丙醇)及丙酮,几乎不溶于乙酸乙酯(乙酸乙酯)。达比加群酯溶解性具有pH 依赖性,pH 低于3 时溶解性较好,体内吸收也会受到制剂微环境的影响,同时,达比加群酯结构并不稳定,酸性、碱性环境中可能发生因酯键水解导致的活性成分降解。因此制剂的处方工艺设计及生产过程控制,可能对制剂的关键质量属性以及体内作用的发挥起到重要的作。1.制备工艺的选择
达比加群酯胶囊多采用微丸包衣或颗粒包衣制备工艺,原研专利对处方及工艺进行了保护,因此国内研究者开发了与原研制剂不同的制备工艺,虽然处方工艺设计不同,但主要目标均一致,一方面为保证仿制制剂的关键质量属性与原研制剂保持一致,一方面为制造一定的体内微环境,以达到与原研制剂生物等效。
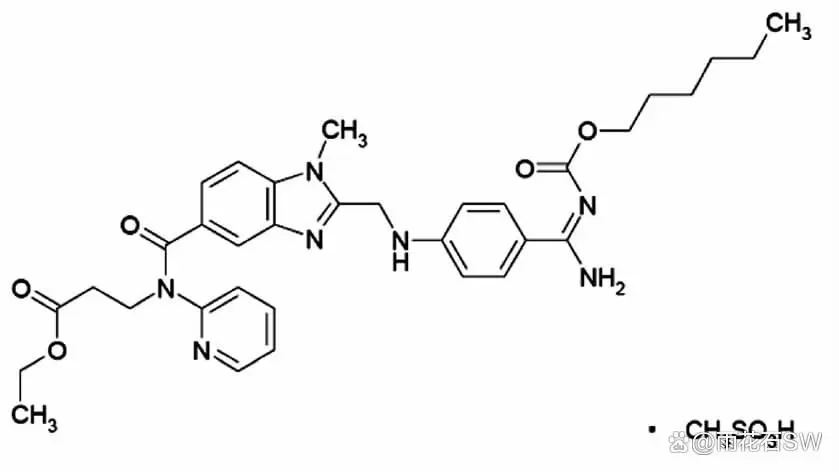
图1:甲磺酸达比加群酯结构式
2.原研制剂制备工艺
根据原研美国上市FDA 审评报告及欧洲药品管理局( EMA) 审评报告,达比加群酯( 活性成分代号BIBR 1048 ) 胶囊制备工艺系将活性成分附着在有机酸丸芯后制得的小丸装填入硬胶囊中,该产品对水分、温度较为敏感,对光不敏感。
专利CN100528157C中说明该产品为有机酸构成的近似球形的芯材料、隔离层和活性成分层构成的多颗粒给药系统,活性成分( 代号BIBR 1048)包裹于有机酸丸芯上,中间通过隔离层将含药层与丸芯分开,其组成见图2。在溶解时,水分渗入到达微丸内部的有机酸溶解后产生“酸性小气候”,即酸性微环境,从而达到活性成分在酸性环境中保持较快溶出的效果,同时隔离层使有机酸与活性成分空间隔离以提高药物的稳定性。
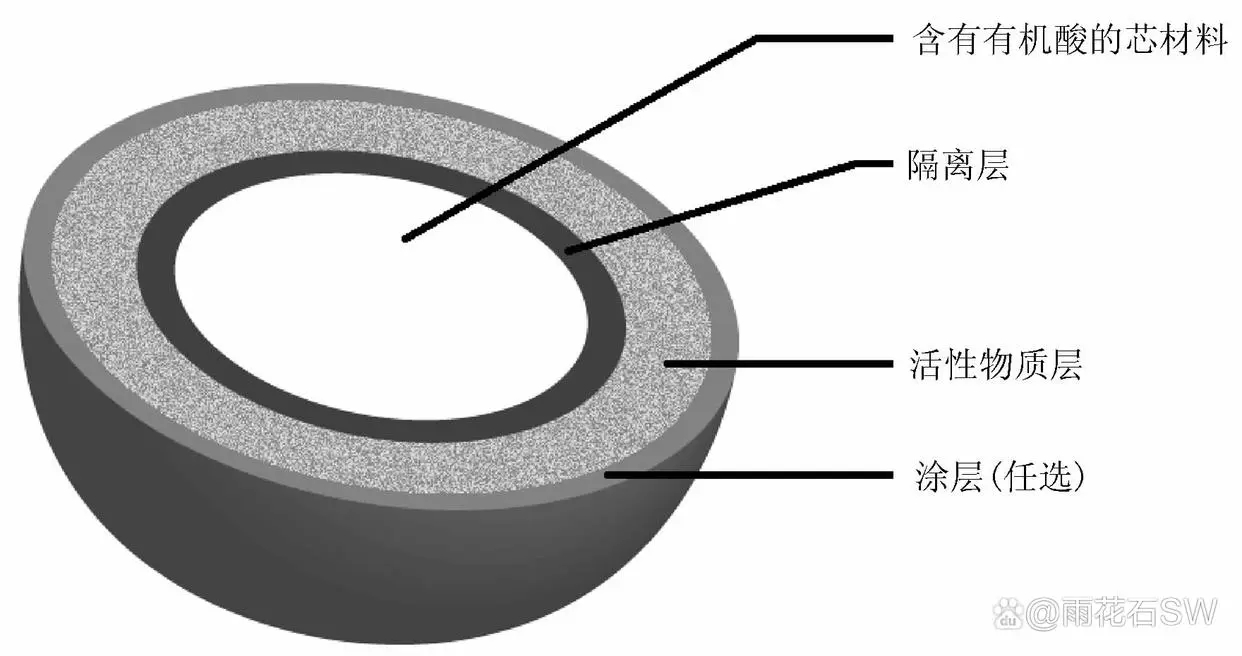
图2:专利CN100528157C 中含药颗粒设计
3.目前国内研究情况
何雄雄等[4]发明了一种含有达比加群酯的药用组合物,包含维生素C(维生素C) 丸芯和活性成分层,且维生素C 丸芯和活性成分层之间依次通过水溶性聚合物层和半透膜层隔离,一方面该隔离层能够在溶出介质中水化,产生孔道使溶出介质渗入,有机酸丸芯溶解并从孔道缓慢释放,产生酸性微环境,从而促进外层药物的溶出; 另一方面,该隔离层能够在溶出过程中包裹丸芯,防止其与外层药物层的分离。该发明能够为活性成分的溶出提供良好的酸性微环境,显著改善产品体外溶出。其组成见图3
维生素C) 丸芯和活性成分层,且维生素C 丸芯和活性成分层之间依次通过水溶性聚合物层和半透膜层隔离,一方面该隔离层能够在溶出介质中水化,产生孔道使溶出介质渗入,有机酸丸芯溶解并从孔道缓慢释放,产生酸性微环境,从而促进外层药物的溶出; 另一方面,该隔离层能够在溶出过程中包裹丸芯,防止其与外层药物层的分离。该发明能够为活性成分的溶出提供良好的酸性微环境,显著改善产品体外溶出。其组成见图3
图3:专利CN105640909 B 中含药颗粒设
专利W0 2016070696 A1[5]中发明了一种达比加群酯制剂制备方法,首先制备球形蔗糖丸芯(蔗糖丸芯),由内至外包衣,第一层为含有活性成分及必要辅料的活性成分层,第二层为水溶性隔离层,第三层为含有有机酸和黏合剂的酸性物质层,第四层为水溶性隔离层,第五层为再次包裹有活性成分和必要辅料的活性成分层,最后一层为含高分子材料隔离层,其组成见图4。
蔗糖丸芯),由内至外包衣,第一层为含有活性成分及必要辅料的活性成分层,第二层为水溶性隔离层,第三层为含有有机酸和黏合剂的酸性物质层,第四层为水溶性隔离层,第五层为再次包裹有活性成分和必要辅料的活性成分层,最后一层为含高分子材料隔离层,其组成见图4。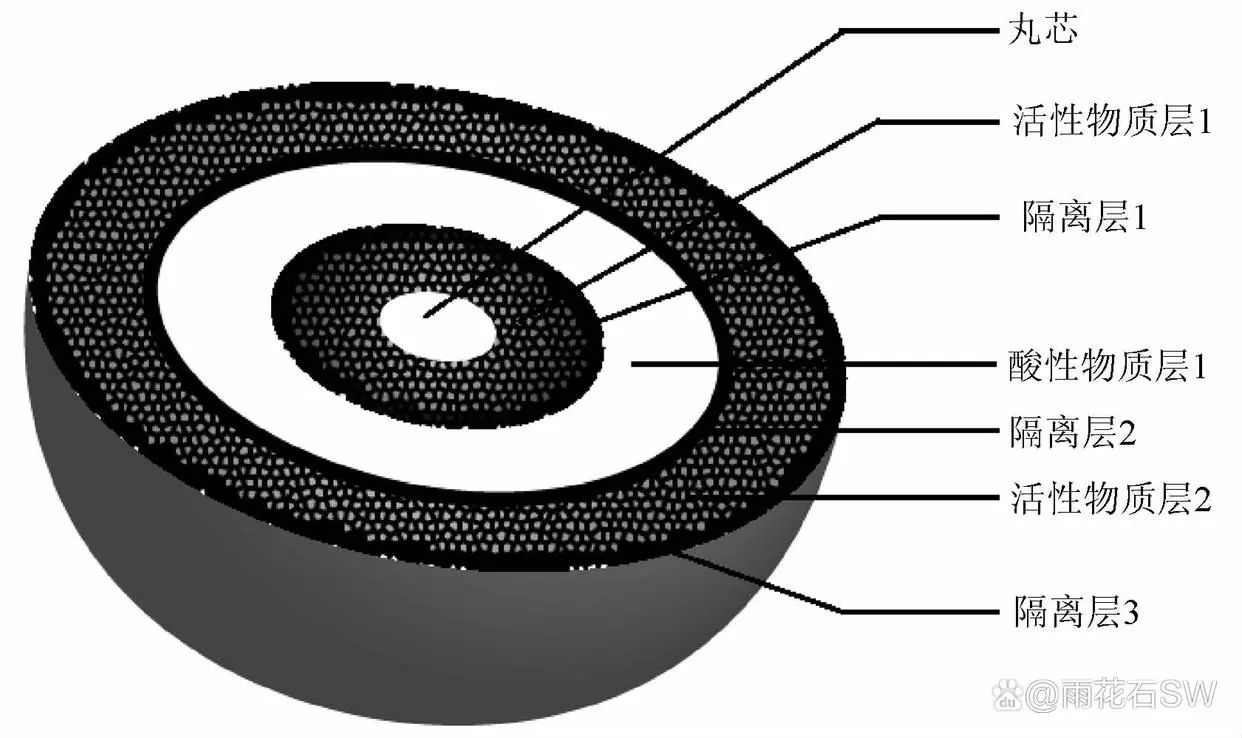
图4:专利W0 2016070696 A1 中含药颗粒
发明人说明该发明的优势为活性成分层位于两层酸性层中间,可以更好的利用水分渗入后酸性物质形成的酸性环境; 另外,制备的产品在中性介质( 例如水) 中具有更好的溶出度; 而且该设计可增加颗粒的耐磨性及贮藏稳定性。王蕊瑞等[6]设计了一种达比加群酯的口服药物组合物,该药物组合物包含了甲磺酸达比加群酯的包衣颗粒和含有酒石酸的包衣颗粒2 个部分,两部分颗粒混合均匀后填充胶囊,其组成见图5。专利说明本发明避免了微丸逐层涂覆的复杂、耗时工艺,制备的胶囊颗粒体积和重量较小,溶出效果较优,同时具有较好的稳定性。
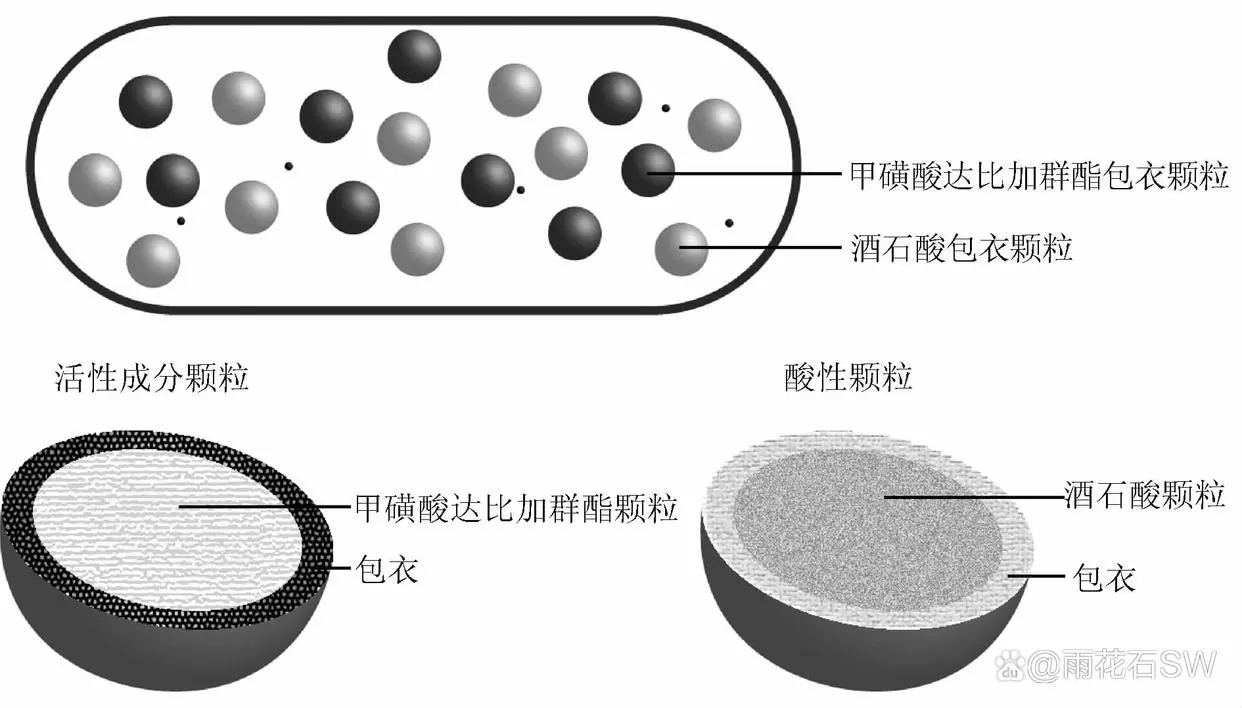
图5:专利CN 110123774 A 中药物设计示
5. 活性成分晶型的控制
根据EMA 及FDA 审评报告,达比加群酯存在多晶型,主要为晶型Ⅰ及晶型Ⅱ,晶型Ⅱ为更为稳定的晶型。原研制剂中活性成分制备工艺得到的主要是Ⅰ晶型、其中混有少量无水Ⅱ晶型,制剂制备工艺及存放过程不会发生转晶;另外,两种晶型的活性成分制备的制剂生物等效,制剂中多晶型现象不会对产品的安全性和功效性产生影响,但原研制剂对晶型Ⅱ的控制比例进行了限度要求。
为了保证与原研制剂体内和体外研究的一致性,仿制制剂一般尽可能采用和原研制剂相同晶型的原料药进行研制开发,重点关注活性成分晶型在制剂制备过程中是否发生变化。达比加群酯胶囊制备工艺一般为流化床上药包衣工艺,制备过程中可能导致活性成分晶型发生改变,为保障仿制制剂批间质量一致性,研究开始时应确认所用活性成分的晶型,研制过程中关注制剂制备过程及稳定性考察过程中是否发生转晶。若所用活性成分为混合晶型( 含有2 种晶型) ,或制备、贮藏过程中发生转晶,但是能够确认晶型的变化不影响产品的生物等效性,那么从工艺控制的角度出发,应考虑建立合理的方法并提供研究数据,证明可以将达比加群酯晶型比例控制在合理的范围内,并保证制剂生产和贮藏过程中批内和批间的一致性。
6活性成分粒度控制
根据EMA 审评报告,原研制剂确定制定了活性成分粒度分布控制限度以确保产品制备过程中质量的一致性。仿制产品采用气流粉碎等微粉化工艺对原料进行预处理,对粒度控制应进行研究,确认不同粒度的活性成分对制剂产品溶出曲线产生的影响。若研究结果显示活性成分粒度分布对制剂产品溶出特性有显著影响,则应对粒度分布制定合理的控制限度,并将活性成分预处理和粒度控制作为制剂生产过程的关键工艺参数。达比加群酯在水中溶解性不佳,如采用水性溶剂配制成包衣液,通常以混悬液的形式存在;如采用其他非水溶剂配制包衣液,可能导致主成分的溶解性不同,进而影响到粒度分布; 因此,处方设计时也应该关注包衣液的配制方法对产品带来的影响。
3 .功能性辅功能性辅料的选择
4 胶囊壳的选择
仿制制剂开发时应关注不同胶囊壳引起的产品溶出特性差异。原研制剂FDA 审评报告及EMA审评报告中说明因活性成分对水解敏感,而羟丙甲纤维素( HPMC) 材质胶囊壳的脆性最小,所以该产品使用HPMC 胶囊壳。根据EMA 审评报告,在体外溶出度试验中,不同胶囊壳材质可能导致前15min 溶出度有差异,其中HPMC 胶囊的释放在15min 内存在约25%延迟,但在30 min 时未观察到差异; 而不同胶囊壳差异对体内影响较小。国内上市的胶囊壳多以明胶(明胶)胶囊为主,若仿制制剂使用的胶囊壳材质与原研制剂不一致,则可能有溶出行为不相似的风险。
明胶)胶囊为主,若仿制制剂使用的胶囊壳材质与原研制剂不一致,则可能有溶出行为不相似的风险。5.活性成分与有机酸的隔离
EMA 审评报告说明酸性环境可提高达比加群酯溶解性,但活性成分在酸性环境及水分存在条件下易于水解,因此制备过程中避免活性成分接触水和酸性环境。因此,在活性成分与酸性化合物之间设计隔离层是保证产品稳定性的基础。综合EMA 及FDA 审批报告中达比加群酯的稳定性研究结果,该类产品处方工艺中隔离层衣膜完整性对于保证活性成分的稳定及给药后含药颗粒中活性成分溶出释放时酸性环境的维持均非常重要。也可以采用分别制丸的工艺将活性成分与有机酸隔离,通常为了避免丸芯表面的接触,不同的小丸也需要包裹隔离层进行保护。无论采用何种制备工艺,研究开发时应对活性成分与酸性化合物的配伍稳定性进行全面的研究,确认是否有产生特异性杂质的风险及采用的隔离层是否可有效阻断两者的直接接触,并提供研究数据说明样品在贮藏过程中有无明显降解,以确认产品贮藏过程中隔离层衣膜的完整性。
6..包衣终点控制及包衣收率研究
流化床包衣的关键工艺参数一般包括流化空气流量、雾化压力、雾化空气流量、进风温度及喷嘴位置等,包衣微丸的粒径、水分及包衣厚度等属性是流化床生产过程中需要监测的重要参数,目前已有多种过程分析技术( process analytical technology,PAT) 应用于流化床包衣工艺控制。国内外研究者多采用流化床包衣技术制备达比加群酯胶囊填充物,一般制备一层或多层包衣含药微丸。微丸包衣工艺终点控制一般采用固定包衣增重或者固定包衣液用量2 种方法。若采用多层包衣的工艺进行含药微丸制备,每层包衣的上药率( 或收率) 是保证产品质量一致性的关键因素。例如,控制隔离层上药率可以对隔离层包衣厚度进行控制,控制活性成分层上药率可以固定酸性化合物与活性成分的总体比例等。流化床包衣是一个连续性较强的制备工艺,衣膜的完整性对包衣性能影响较大,尤其是隔离层衣膜的完整性对达比加群酯胶囊的稳定性十分重要,而制备过程中小丸包衣增重较难实际测量,推荐采用固定包衣液用量的方法进行达比加群酯含药微丸各层包衣终点控制。应通过处方工艺研究及多批次工艺验证确定包衣液用量,设定每层包衣上药率的合理控制范围。另外,各层包衣喷液过程中会发生包衣液损失,可能影响到各层包衣收率及终产品含量,尤其是含药层上药包衣,应在工艺研究验证过程中进行充分的研究验证,评估管路残留、过滤不完全、容器吸附等风险因素是否导致隔离层或活性成分层上药不完全。
7. 关键工艺参数设置和中间产品质量控制
仿制药研究过程中关键工艺参数选择及中间产品控制标准制定均应经过合理的评估。对于达比加群酯胶囊,生产开发过程中应对上药包衣、含药微丸( 或颗粒) 混合、胶囊灌装工序参数进行研究确认,根据风险评估及工艺验证结果,将对产品质量影响较大的工艺参数定为关键工艺参数,并进行严格控制。达比加群酯胶囊的中间产品一般包括各层包衣后微丸( 或颗粒) ,各层包衣厚度及增重可能影响终产品的溶出特性,应在处方研究及工艺参数确认过程中,拟定合理的中间产品控制指标及标准,并根据研究结果,将可能影响成品质量的关键控制项目( 如包衣后微丸的包衣上药收率、溶出度及溶剂残留等) 定入中间产品质量标准,制定合理的控制限度,并根据中间产品稳定性考察结果制定合理的存储条件及使用有效期。另外,若将活性成分或功能性酸性成分配制为包衣液进行微丸( 或颗粒) 包衣,应对包衣液的稳定性进行考察,评估是否有发生混悬液絮凝或者活性成分降解的风险,确认包衣过程中包衣液的稳定性及使用时限。
8 .批量放大
产品研制过程中可能遇到因批量放大需进行设备变更的情况,生产工艺若发生变更应以拟定的包衣上药率、成品溶出曲线特性等考察项目为指标,参考变更相关指导原则(指导原则),对变更后设备及相关工艺参数进行全面的研究验证,以保证批量放大后产品的关键质量属性符合拟定要求。另外,若产品制备工艺为分亚批制备含药微丸( 或颗粒) 后混批进行胶囊灌装,分多个亚批进行微丸( 或颗粒) 制备或上药包衣,应在研究过程中重点对不同亚批包衣微丸( 或颗粒) 的粉体学性质进行考察,以实现微丸( 或颗粒) 亚批间的质量一致性。同时,应根据商业化规模实际生产情况,确定包衣微丸( 或颗粒) 亚批中间产品的合理存储时限,确认产品生产过程中质量的稳定性。
指导原则),对变更后设备及相关工艺参数进行全面的研究验证,以保证批量放大后产品的关键质量属性符合拟定要求。另外,若产品制备工艺为分亚批制备含药微丸( 或颗粒) 后混批进行胶囊灌装,分多个亚批进行微丸( 或颗粒) 制备或上药包衣,应在研究过程中重点对不同亚批包衣微丸( 或颗粒) 的粉体学性质进行考察,以实现微丸( 或颗粒) 亚批间的质量一致性。同时,应根据商业化规模实际生产情况,确定包衣微丸( 或颗粒) 亚批中间产品的合理存储时限,确认产品生产过程中质量的稳定性。
