6.2.5 文件管理
企业开展信息化工.作时,以下关于文件管理的GMP 要求,可以考虑使用信息化系统管理。
法规要求
药品生产质量管理规范(2010年修订)
第一百五十一条 企业应当建立文件管理的操作规程,系统地设计、制定、审核、批准和发放文件。与本规范有关的文件应当经质量管理部门的审核。
第一百五十三条 文件的起草、修订、审核、批准、替换或撤销、复制、保管和销毁等应当按照操作规程管理,并有相应的文件分发、撤销、复制、销毁记录。
第一百五十四条 文件的起草、修订、审核、批准均应当由适当的人员签名并注明日期。
第一百六十三条 如使用电子数据处理系统、照相技术或其他可靠方式记录数据资料,应当有所用系统的操作规程;记录的准确性应当经过核对。
使用电子数据处理系统的,只有经授权的人员方可输入或更改数据,更改和删除情况应当有记录;应当使用密码或其他方式来控制系统的登录;
关键数据输入后,应当由他人独立进行复核。
若上述部分实现由系统管理,一般体现为:
• 系统应支持对文件内容和文件格式的模板进行配置管理;支持对起草、核对、批准、发放等工作流程进行配置管理;
• 当对文件执行相关操作,系统应支持留下电子记录;
• 工作流的确认、核对、批准、驳回或变更数据等关键步骤执行时,需要电子签名。
法规要求
药品生产质量管理规范(2010年修订)
第一百五十八条 文件应当定期审核、修订;文件修订后,应当按照规定管理,防止旧版文件的误用。分发、使用的文件应当为批准的现行文本,已撤销的或旧版文件除留档备查外,不得在工作现场出现。
若上述部分实现由系统管理,一般体现为:
• 对文件进行版本控制,所有使用文档应当在有效期内,不允许同时有两个版本文件在系统中处于可使用状态。
法规要求
药品生产质量管理规范(2010年修订)
第一百六十二条 每批药品应当有批记录,包括批生产记录、批包装记录、批检验记录和药品放行审核记录等与本批产品有关的记录。批记录应当由质量管理部门负责管理,至少保存至药品有效期后一年。
第一百六十三条 用电子方法保存的批记录,应当采用磁带、缩微胶卷、纸质副本或其他方法进行备份,以确保记录的安全,且数据资料在保存期内便于查阅。
若上述部分实现由系统管理,一般体现为:
•系统应支持对批生产记录、批包装记录、批检验记录和药品放行审核记录的记录和保存;
•系统应支持企业信息化部门数据备份管理策略,满足法律规范对于文件保存期限的管理要求,例如批记录需要满足“至少保存至药品有效期后一年”的要求。
实施指导
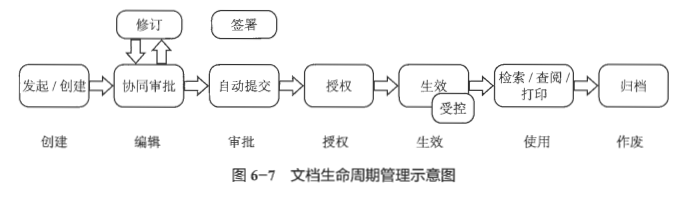
GMP文件管理一般由文档管理系统(DMS)实现信息化管理。对于批生产记录、批包装记录、批检验记录等当然也可以由 MES、LIMS 等系统实现管理。
DMS 是对文档从创建、审批、发布、培训、再修订到作废的生命周期进行管理,帮助企业确保文档的一致性和合规性。对文档的权限控制保证了文档只能被授权的用户查看,确保了文档的安全性。
DMS 基本功能包括:
• 文档模板和内容模板的创建;
• 业务流程的在线审批和文件的变更修订;
• 文件的打印、分发和回收;
• 文件的归档和作废;
• 自动生成文件复审日期和复审计划;
• 其他功能如基础信息管理、日志管理、权限管理,以及相关符合数据可靠性要求的功能。