

嗯,上周断更,没发出去,今天继续,上图:
一、【基本概念】
l批记录(Batch record):用于记述每批药品生产、质量检验和放行审核的所有文件和记录,可追溯所有与成品质量有关的历史信息。批记录包括批生产记录、批包装记录、批检验记录、药品放行审核记录和其他与本批产品有关的记录文件。
l批生产记录(ManufacturingRecord,Batch production record):依据现行批准的工艺规程制定,用来追溯该批产品生产历史以及相关的质量情况。按照FDA CFR 211,批生产记录的空白模板是主生产记录(Master production record)
l批包装记录(Package record):依据工艺规程中与包装有关的内容制定,用来追溯该批产品包装操作以及相关的质量情况。
l批检验记录(Test record,Batch control record):用来追溯该批产品的检验和质量状况。按照FDA CFR 211,批检验记录的空白模板是主检验记录(Master control record)。
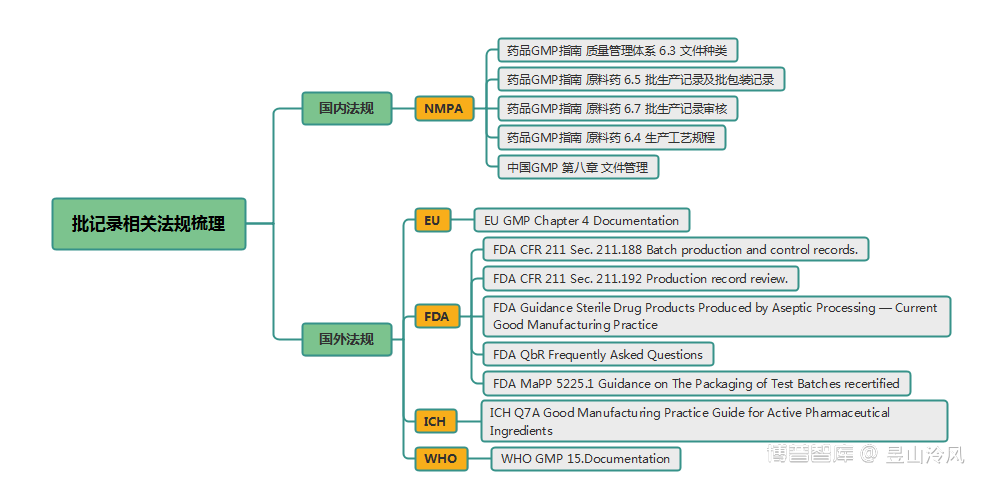
二、相关法规
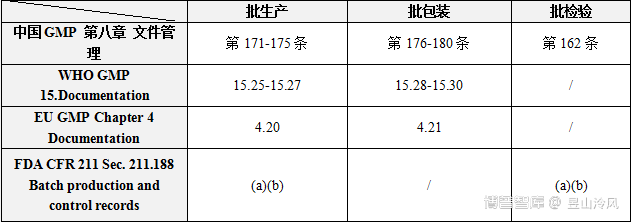
【中国GMP 第八章 文件管理】
第一百六十二条 每批药品应当有批记录,包括批生产记录、批包装记录、批检验记录和药品放行审核记录等与本批产品有关的记录。批记录应当由质量管理部门负责管理,至少保存至药品有效期后一年。
质量标准、工艺规程、操作规程、稳定性考察、确认、验证、变更等其他重要文件应当长期保存。
【FDA CFR 211 Sec. 211.188 Batch production and control records】
Batch production and control recordsshall be prepared for each batch of drug product produced and shall include complete information relating to the production and control of each batch. These records shall include:
(a) An accurate reproduction of the appropriate master production or control record, checked for accuracy, dated, and signed;
(b) Documentation that each significant step in themanufacture, processing,packing, orholdingof the batch was accomplished, including:
(1) Dates;
(2) Identity of individual majorequipmentand lines used;
(3) Specific identification of each batch ofcomponentorin-process material used;
(4)Weightsand measures of components used in the course of processing;
(5)In-processandlaboratorycontrol results;
(6) Inspection of thepackagingandlabelingarea before and after use;
(7) A statement of the actual yield and a statement of the percentage of theoretical yield at appropriate phases of processing;
(8) Complete labeling control records, including specimens or copies of all labeling used;
(9) Description of drug productcontainers and closures;
(10) Anysamplingperformed;
(11) Identification of the persons performing and directly supervising or checking each significant step in the operation, or if a significant step in the operation is performed by automated equipment under211.68, the identification of the person checking the significant step performed by theautomated equipment.
(12) Any investigation made according to211.192.
(13) Results of examinations made in accordance with211.134
【FDA CFR 211 Sec. 211.192 Production record review】
Alldrug productproduction and controlrecords, including those forpackagingandlabeling, shall be reviewed and approved by thequality control unitto determine compliance with all established, approved writtenproceduresbefore a batch isreleasedordistributed. Any unexplained discrepancy (including a percentage of theoretical yield exceeding the maximum or minimum percentages established in master production and control records) or the failure of a batch or any of its components to meet any of itsspecificationsshall be thoroughly investigated, whether or not the batch has already been distributed. The investigation shall extend to other batches of the same drug product and other drug products that may have been associated with the specific failure or discrepancy. A written record of the investigation shall be made and shall include the conclusions and followup.
【EU GMP Chapter 4 Documentation】
Retention of Documents
4.11 Specific requirements apply to batch documentation which must be kept for one year after expiry of the batch to which it relates or at least five years after certification of the batch by theQualified Person, whichever is the longer. For investigational medicinal products, the batch documentation must be kept for at least five years after the completion or formal discontinuation of the last clinical trial in which the batch was used. Other requirements for retention of documentation may be described in legislation in relation to specific types of product (e.g. Advanced Therapy Medicinal Products) and specify that longer retention periods be applied to certain documents.
三、相关指南
【(无菌生产的批记录与检验)】
The requirement for review of allbatch recordsand data for conformance with writtenprocedures, operating parameters, and productspecificationsprior to arriving at the finalreleasedecision for an aseptically processed product calls for an overall review of process and system performance for that given cycle of manufacture. All in-process andlaboratory controlresults must be included with thebatch recorddocumentationin accordance with section211.188. Review ofenvironmentaland personnelmonitoringdata, as well as other data relating to acceptability of output from support systems (e.g.,HEPA / HVAC,WFI, steam generator) and proper functioning of equipment (e.g., batch alarms report; integrity of various filters) are considered essential elements of the batchreleasedecision.
【(批记录在申报文件中的位置)】
Include batch records for the ANDA batch and proposed commercial batches in section 3.2.R.1.P.1 or 3.2.P.3.3 . The ANDA checklist of 10/10/2006 recommends 3.2.P.3.3, but according to the 2001 Guidance for Industry M4Q: The CTD - Quality (http://www.fda.gov/cder/guidance/4539Q.htm
【(申报批包装的一般原则)】
5. The Packaging and Labeling Sections of thebatch recordshould contain complete records for the packaging andlabelingoperations, including drug product and label reconciliation.
6. The Packaging Section of thebatch recordshould include a summary table of packaging information describing the container/closure system, the total number of containers packaged and the quantity disbursed, and the destination of all disbursements of the packaged product.
四、归纳
批记录的风险点

参考药品GMP指南 质量管理体系 6.3 文件种类。不同记录审核内容示例

参考药品GMP指南 原料药 6.7 批生产记录审核
六、【实例分析】
工艺规程与批记录母本对原料药生产工艺规程的要求,和Q7中对于MP&CR(Master Production and Control Record,主生产和控制记录)的相关要求相似。在Q7中,MP&CR就是指的批记录的母本,包括批生产记录和批包装记录的母本。某公司希望将批记录的母本和生产工艺规程合并,在批记录母本修订时,参照法规中对生产工艺规程所要求的所有信息,将生产工艺规程所需要信息和批记录母本中已经有的信息进行比较,然后把生产工艺规程中要求的信息全部加入批记录母本。这样使得批记录的母本满足了生产工艺规程的所有要求,两个文件合二为一。药品GMP指南 原料药 6.4 生产工艺规程、ICH Q7A Good Manufacturing Practice Guide for Active Pharmaceutical Ingredients

