


写在前面的话:该部分主要是基础理论,关于RSE的历史沿革有助于我们理解制定标准品的考虑。对于LBS的结构及致热相关机制有助于理解如何制定LER研究策略。
正文
2016年3月在德克萨斯州圣安东尼奥举行的第一次PDA LER研讨会,并将该次达成一致的提议发表于2016年9月PDA Letter:““PDA’s LER Task Force Holds its First Workshop””(67)。
基于相关科学研究和论证工作过程所获取的额外信息,本技术报告阐述了使用鲎试剂(LAL)检测特定制剂中内毒素的适用性问题。本章节论述了美国法规有关LAL试验的规定、用于LAL试验的适宜标准品、生产过程中的的微生物控制以及内毒素对人类健康的影响。
6.1 Regulatory Framework法规框架
美国FDA法规要求对无菌或无热原的药品每批都进行无菌和热原检查(21 CFR 211.167)。这些法规要求,特别是21 CFR 211.160(b),旨在确保最终产品符合科学合理且相应的质量与纯度标准(68)。生物制品的具体法规(21 CFR 610.13(b))规定,“拟用于注射用产品的每批终容器内的产品都应通过静脉注射到家兔体内的方式检测热原……”(69)。生物制品最初必须采用美国药典<151>-热原检查(药典)或21 CFR 613(b)-纯度(监管)方法进行检测,以确认制剂本身无热原;根据21 CFR 610.13(b)规定,毒素或类毒素产品无需进行该检测。在证明制剂分子不致热时(Once the molecule is shown to be nonpyrogenic),可使用相对更灵敏、可靠的USP细菌内毒检查法代替USP<151>热原试验进行生物制品的放行。
注释1:最后一句话说的molecule应该是指制剂不仅包括有效成分还包括杂质成分致热,此时仅是内毒素检查合格仍致热,那么就需要使用家兔进行热原检查。
经美国药典(USP<85>)、欧洲药典(Ph. Eur.2.6.14)、WHO(国际药典第3.4章)和日本药典(JP 4.01) 协调一致的药典方法易受到检测干扰(2-6)。可通过样品稀释不超过最大有效稀释倍数(MVD)或其他经过验证的样品制备方法来减轻消除检测干扰。当这些方法很难消除干扰时,可以开发和使用符合21 CFR 610.9的替代方法(70)进行检测。该法规要求申请人提供证据证明新的或改良的标准方法(法规或药典)与标准方法等效(equivalent to the standard method)。这与美国药典凡例的章节6.30一致,必须对替代方法或程序进行全面验证,且必须在允许限度(根据具体情况确定的)内与药典方法或程序相当 (71)。如果改进或等效方法不能缓解检测干扰,则应使用家兔热原试验或其他等效方法(12)。
在建立终产品的内毒素质量标准时,该限度应低于在人体中产生热原反应所确定的限度(例如,静脉内给药5 EU/kg/小时或鞘内给药0.2 EU/kg/小时)。此外,在建立最终内毒素质量标准时,应考虑内包装组件、处方辅料(formulation excipients)、注射用水(WFI)和散装原料药(bulk drug substance)的内毒素最大理论贡献,以确保不超过理论内毒素限度(72,73)。
注释2: 研发阶段进行内毒素质量研究时,原辅料和注射用水不会被忽略,但是内包材很容易被忽略。其次内包材的内毒素贡献如何计算就是一个麻烦,可通过使用无热原内包材来实现(如隧道烘箱灭菌),根据接触面积来逆推也是一种方法。
6.2 Microbial Control During Manufacturing and Product Quality药品生产和至产品质量控制过程中的微生物控制
大多数治疗性蛋白产品来源于天然或重组微生物或哺乳动物细胞表达系统。这些产品和工艺中间体采用复杂的多工序生产时存在被来源于原材料、人员和生产环境(生物负载、内毒素的外源因子的潜在来源)的微生物污染的风险。工艺过程和产品中间体中污染的微生物可能会增殖并产生蛋白酶和其他副产物,进而影响最终制剂的安全性、纯度和效价。例如,微生物蛋白酶可以在生产过程和货架期内剪切和降解蛋白质产品,影响产品纯度、免疫原性和效价。其他微生物副产物,如内毒素和其他病原体相关分子模式(PAMP),可能影响安全性(74)。生产过程的各个步骤都必需进行微生物控制,以确保终产品符合既定的安全性、纯度和效价标准。
全面的微生物控制策略对于安全有效产品的生产至关重要。全面控制策略包括设施设备的设计和维护即将污染和交叉污染风险的风险降到最低。这些设施可能包含通过HEPA过滤空气、压差和换气来将污染风险降到最低的分级区域。应对这些设施进行适当的清洁和环境监测以确保微生物的控制。设备被作为封闭系统进行运行应尽可能将微生物污染风险降到最低,并进行适当的清洁、消毒和(或)灭菌。对于产品暴露于环境的操作,应当有额外的适当控制措施。此类控制可能包括人员穿着适当的工作服使用局部层流罩和经常使用使用0.2 μm过滤器过滤中间产品以去除可能的微生物污染物。应当在工艺中最易受微生物污染的点进行生物负载和内毒素检测,并及时监控记录结果以确保微生物被有效控制。开放操作、人员频繁操作或放置时间超过24小时时微生物污染的风险很高,应进行密切监测和控制这些操作。微生物的存在可以使用生物负载试验进行检测和定量,生物负载试验可反映工艺或产品中被污染的微生物数量和类型相关信息。在某些情况下,可使用生物负载检测结果作为终产品中毒素和非内毒素病原分子相关模式的替代检测(75)。这些替代和快速生物负载检测方法对实时进行工艺监测和控制很有帮助。然而,这些方法尚未得到广泛实施。相比之下,可以在2小时内完成检测的内毒素检查可在过程控制时(in-process)更及时提供微生物控制状态的信息。最后,细菌内毒素检查可提供额外的保障,即最终产品的总内毒素负载低于致热阈值。
6.3Endotoxin Structure内毒素结构
内毒素或脂多糖(LPS)是革兰氏阴性菌外膜的主要成分;它由O-抗原多糖(O-antigen polysaccharide)、核心寡糖(core oligosaccharide)和类脂A(lipid A)组成(76)。类脂A是LPS(内毒素)的生物活性部分,通过toll样受体4/髓系分化蛋白2(TLR4/MD2)激活先天性免疫系统,TLR4/MD2可由单核细胞、巨噬细胞和血小板等多种细胞表达 (77,78)。此外,类脂A负责将LPS锚定在革兰氏阴性菌的外膜内,是LPS的保守分子组分。
注释3:细胞外膜在革兰氏阴性菌的表层,有着一个多重结构的细胞壁。在细胞质膜的外面,有着一层主要由肽聚糖组成的细胞的细胞壁,细胞壁外还有一层由蛋白质、磷脂质、脂多糖形成的膜层,为了与里面的细胞质膜相对应,特称此膜层为细菌外膜。
注释4:Toll样受体(Toll-like receptors, TLR)是参与非特异性免疫(天然免疫)的一类重要蛋白质分子,也是连接非特异性免疫和特异性免疫的桥梁。TLR是单个的跨膜非催化性蛋白质,可以识别来源于微生物的具有保守结构的分子。当微生物突破机体的物理屏障,如皮肤、粘膜等时,TLR可以识别它们并激活机体产生免疫细胞应答。
注释5:O多糖位于脂多糖最外层与环境相接触,是革兰阴性细菌的菌体抗原(O抗原),典型的O多糖由20~40个重复单位组成,每个重复单位由3~7个糖分子构成,因为各菌种的O多糖种类和排列顺序均不相同,因此,O多糖又称为特异性多糖,是革兰阴性细菌分类的重要依据。0多糖的结构是LPS 中最易发生变化的部分,并可引起特异性的免疫反应。核心多糖位于O多糖与脂类之间,可分为连接O多糖的外核部分和连接脂类的内核部分。核心多糖相对稳定,当LPS缺乏O 多糖时,它也能诱导抗体产生,但抗体的影响作用较小。内核部分含庚糖和2-酮基-3-脱氧辛酸(KDO)两种特殊的糖类分子,它们在LPS结构中起连接多糖与脂类的作用。脂类主要指类脂A,位于LPS内层由2个葡萄糖胺、磷酸盐和一定量的脂肪酸组成,为LPS中最稳定的部分。目前认为在LPS结构中只有类脂A及KDO部分具有毒性,此结构部分具有激活机体免疫系统的作用。
在大多数革兰氏阴性菌中类脂A的生物合成是一个保守且有序的过程。然而,一些革兰氏阴性菌缺乏这种生物合成途径(76,79,80)。Christian Raetz的实验室表征并阐明了大肠杆菌类脂A生物合成途径是由9个酶促反应组成。最重要的是,大肠埃希菌中的内毒素生物合成途径是典型的类脂A代表结构,由6条脂肪酸链以不对称模式和2个未修饰的磷酸基团连接在糖骨架上(76,80,81)。
许多革兰阴性菌也具有改变其类脂A结构的酶机制,导致来源于不同僧问题的类脂A结构的多样性。此外,研究证明类脂A的结构异质性取决于革兰氏阴性微生物的生长和环境条件(例如,pH值、温度、二价阳离子浓度等)。这些修饰通常涉及酰基链数量变化、酰基链长度的变化和/或磷酸基团的修饰(79)。大肠杆菌的己烯酰化类脂A结构(图6.3-1)是一种已知的有效免疫调节剂,结构研究数据表明类脂A部分的特定部分与TLR4/MD-2相互作用可增强先天免疫反应(77,82,83)。并非所有的内毒素结构都能引起这种免疫反应,如革兰氏阴性菌幽门螺杆菌仅有4条脂肪链的内毒素结构(84,85)。确切的说,研究表明幽门螺杆菌的四酰化类脂A结构的效力比大肠埃希菌的类脂A效力低1000倍(86-89)。此外,Loppnow 等人的研究(见第6.7节)表明,白细胞介素-1(IL-1)的产生取决于LPS的结构(图6.3-2)(90)。最重要的是,革兰氏阴性菌中类脂A种类的多样性和异质性强调了使用充分表征的标准品进行制剂内毒素检测的重要性,例如,从大肠埃希菌中分离的参比标准品内毒素(RSE)。此外,在生产环境中经常发现的的各种革兰氏阴性菌的类脂A结构多样性还未得到表征。
注释6:在革兰阴性杆菌生长繁殖过程中,内毒素也不断从细胞壁上脱落下来并释放到周围的介质中去。
肠道中寄存着数目庞大、种类繁多的细菌,其中的大肠杆菌等革兰阴性菌不断向肠腔中释放内毒素。因此,肠腔不仅是“储菌所”,也是“内毒素库”,肠腔中的内毒素含量足以导致宿主死亡,但由于肠黏膜具有良好的屏障功能,故生理情况下仅有少量内毒素向肠外移位,而且这些内毒素很快被肝脏所破坏。
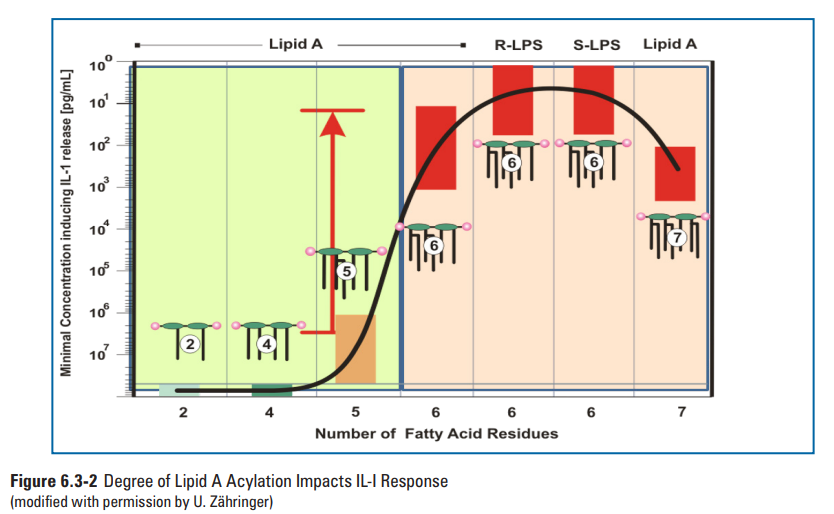
图6.3-2白介素-1与类脂A结构关系(图片来源于PDA TR82)
6.4Limulus Amebocyte Lysate (LAL) Assay鲎试剂分析
1956年,Bang观察到美洲鲎(马蹄蟹)因感染革兰氏阴性菌导致血淋巴(血液)凝固(91)。随后,Levin和Bang证实凝血激活是对细菌内毒素(革兰氏阴性菌外膜的主要成分)的直接免疫反应(92-94)。整个凝血机制在变形细胞(鲎血液中唯一的一种细胞类型)中进行(93)。经不断研究,对鲎变形细胞裂解物进行洗涤纯化后可用于微量细菌内毒素的测定(94)。观察发现,内毒素导致变形细胞裂解物的凝血的反应依赖于温度和pH值,对丝氨酸和半胱氨酸蛋白酶抑制剂敏感,表明该凝血系统本质上是一个酶促反应(95)。因此, LAL测定法经评估后在临床上应用与诊断败血症 (96,97)。
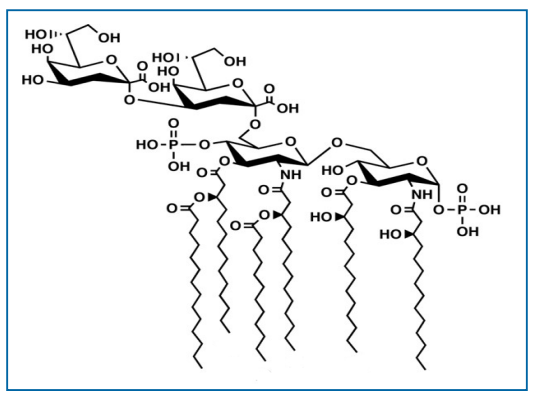
图6.3-1大肠埃希菌类脂A结构图(图片来源于PDA TR82)
因LAL是一种血液制品,美国FDA决定编写关于其制备和使用的法规(98,99)。1973年1月12日,监管机构宣布其计划许可LAL,并声明由于鲎试剂变异性LAL测定法不能用作人用药物的最终热原测定(100,101)。不过,该检测可用于检测药物生产过程中的内毒素检测。1973年9月18日确立了LAL许可的其他标准,认为除非使用参比内毒素参考品进行了鲎试剂灵敏度试验,否则LAL无法获得许可。具体来说,鲎试剂应对0.1 ng内毒素参考品(reference endotoxin)产生阳性反应,对0.0125 ng内毒素参考品产生阴性反应。1974年11月13日,生物制品管理局(Bureau of Biologics现为生物制品评价和研究中心或CBER)认识到细菌内毒素是灭活流感疫苗不可避免的污染物。因此,在21 CFR 610.11中增加了一项要求,即每种流感疫苗均需要对照FDA提供的参比标准品制剂进行内毒素检测(102)。尽管未专门要求LAL方法,但大多数生产商使用LAL试验测定内毒素浓度(103)。
在对生产工艺进行全面审查(包括生产设施和操作规范)后,于1977年批准了第一种鲎试剂的许可。根据许可证,对每批鲎试剂都应进行检测以核实其标识灵敏度(claimed sensitivity )(98)。随着生产技术的标准化和改进,1977年11月4日,FDA批准LAL可用于生物制品和医疗器械中家兔热原试验(RPT)的替代方法(104)。
1980年1月18日,FDA发布了关于LAL可用于代替药品(非生物制品)家兔热原测试的公告(105);1983年3月29日发布了另一份LAL可用于医疗器械和生物制品的草案(106)。1987年12月,发布了可使用LAL试验作为人用和动物用注射药物、生物制品和医疗器械最终产品内毒素检查(代替家兔热原法使用条件)的指南(107)。2011年6月撤销1987年发布的指南,并于2012年6月替换为行业指南:Pyrogen and Endotoxins Testing: Questions and Answers。
FDA许可的在役的(active FDA-licensed)LAL生产商为:
•Associates of Cape Cod, Inc. (License #0700)
•Baxter Healthcare Corporation (License #0140)
•Charles River Endosafe, a Division of Charles River Laboratories, Inc. (License #1197)
•Lonza Walkerville, Inc. (License #1775)
•Wako Chemicals USA, Inc. (License #1762)
6.5Endotoxin Reference Standards内毒素参比品
早在1950年世界卫生组织(WHO)会议上,我们就了解了制备和界定生物参考标准品的困难和特殊性。20世纪50年代初,干燥菌(通常为痢疾杆菌)被用于家兔热原法的标准化和控制。经过四年的辩论和讨论,WHO确定以纯化后的内毒素作为热原参比标准品 (108)。在这样做的过程中,本组织开始了基于单一来源的纯化、表征的大分子制备具有普遍性、简化和标准化的生物标准品的趋势,这反映在自1989年以来制定三个国际标准的方式中。
由于基于美洲鲎的方法本质上是生物测定法,鲎试剂本质上是可变的。因此,需要用耐用、可靠的标准内毒素来确定每批LAL的灵敏度。这些研究的早期贡献者指出RSE的理想特征应为:
1)Well-known and pyrogenic in nature众所周知的热原性
2)Well-characterized特征明确
3)Capable of producing a positive febrile response in rabbits (for RPT correlation)能够在家兔中产生性发热反应(用于家兔热原测试相关性)
4)Stable over a number of years数年内保持稳定
5)Produced in sufficient quantity足量生产
6)Reliable and reproducible可靠且可重现
因此,RSE在制备时是均匀的,容易重悬,并且不易被稀释中使用的试管或容器吸附(109)。
6.5.1U.S. FDA Development of Reference Standard Endotoxins U.S.FDA开发的内毒素标准参比品
美国FDA于1972年开始努力建立可靠的参考标准品,在药物局Dr. D.B. Castor的实验室中,将一批肺炎克雷伯菌内毒素(批次1b)冻干(在0.1%人血清白蛋白中)并使用数年。在不同实验室中观察到批次1b的热原测试结果存在差异。尽管没有直接证据表明内毒素制备液是批次1b变异性的原因(更可能原因使用家兔进行热原检查引起的),但使用更科学合理的标准品代替批次1b。从来源于蒙大拿大学J.a. Rudbach博士的大肠埃希菌O113:H10:K(-)提纯得到的LPS被作为了RSE。由FDA制备相关批次,并作如下制定:EC-1由1975年的大肠杆菌生产(200瓶规模的生产),EC-2由1976年的更大规模(1500瓶)后续生产。在内毒素专家的敦促下,将内毒素单位(EU)为EC-2赋值,即1.0 EU = 0.2 ng EC-2。因此,未来不同效价的内毒素制剂可以直接相关,而不依赖于质量。因为“EU”当前用于标识LAL对内毒素的灵敏度,如果拟定的内毒素标准品经过化学纯化和充分特性鉴定并且其效价与RSE相关,那么内毒素来源的实际选择并不重要。FDA使用由美国药典委员会(USP,见以下章节)分销的大肠埃希菌(EC)内毒素。
由于已知人血清白蛋白(HSA)可结合纯化的LPS,FDA试图生产不含HSA的参比标准品(EC-3和EC-4),但这些批次的效价不满足需求(58)。FDA随后与Mallinckrodt制药公司签订合同,准备生产大批量不含人血清白蛋白的RSE来替代EC-2。在最终制备中使用乳糖和聚乙二醇作为稳定剂代替人血清白蛋白。该批次共生产了50,000瓶:一半给FDA CBER(命名为EC-5),另一半给USP(将其称为批次F,见下文)。当前RSE制剂(EC-6)由伦敦国家生物制品检定所开发,与EC-5的稳定剂相同;由美国药典、日本药典和欧洲药品质量管理局(EDQM)分销,批号为G(见下表6.5.1-1)。
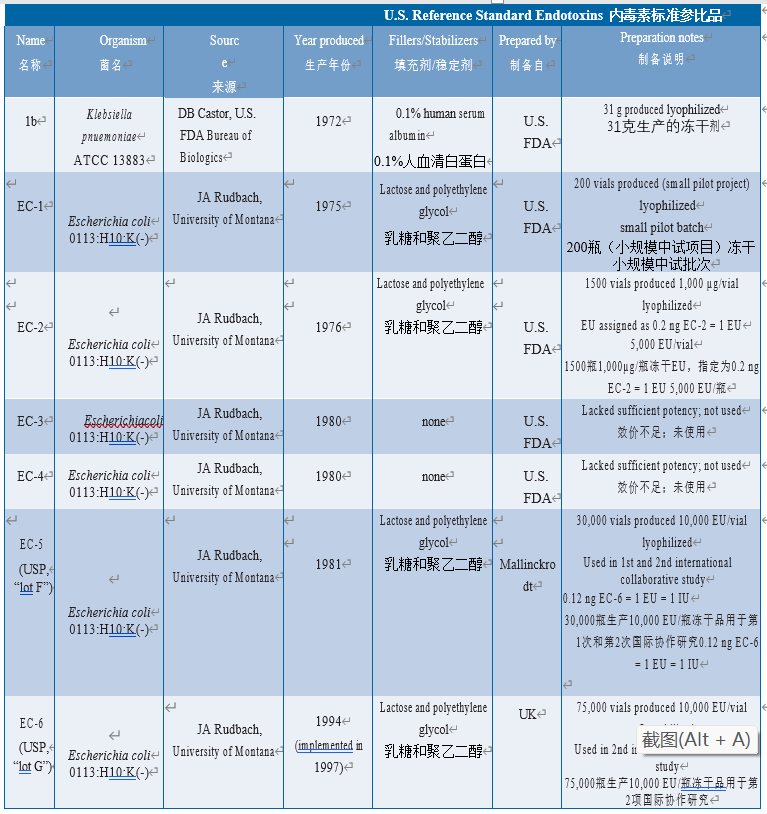
表 6.5.1-1 U.S. Reference Standard Endotoxins development history(图片仅用于学习交流)
6.5.2Harmonization of International Reference Standard Endotoxins内毒素标准参比品的国际协调
国际上大量在用不同内毒素标准制剂给医药行业带来巨大的挑战和混乱。考虑到内毒素检查作为关键安全性指标的重要性,需要全球协调的参比物质,以降低关键检测参数的变异性水平。为此,由美国FDA将来自菌株0113:H10:K(-)的纯化原液材料捐赠给美国药典,并用作WHO内毒素国际标准品(1989年第1个国际标准品、1996年第2个国际标准品和2012年第3个国际标准品)以及欧洲药典的起始物料。BRP制备液3和4,以及从批次F开始的USP参比标准品。WHO国际标准品由基于大型国际研究的WHO生物制品标准化专家委员会(ECBS)委托和批准,以确立一致的效价。第一种IS含2 μg内毒素(与USP/FDA STD EC5相同的原液)+ 3 mg海藻糖/安瓿。第2和第3代IS含1.2 μg内毒素/瓶,用乳糖和聚乙二醇配制。ECBS仅根据凝胶法试验数据标定1st IS,并使其成为仅用于凝胶法的标准品。在使用第二个IS的情况时, WHO、USP/FDA和EDQM标准得到了更好地协调。自第二国际标准品发布以来,欧洲药典和美国药典标准品的处方和灌装均与国际标准品相同,并且在同一研究中确定了其效价。所有材料均已在英国国家生物标准与检定所(NIBSC)配制和灌装。自1996年第2次国际标准品发布以来,已经确定了1 IU = 1 EU的单位等效性(表6.5.2-1)(64,112,113)。
日本国家参考内毒素历来是基于不同菌株的材料,与上述材料不一致。最近,人们希望建立一种与现有国际、欧洲和美国参考标准高度相当的材料。
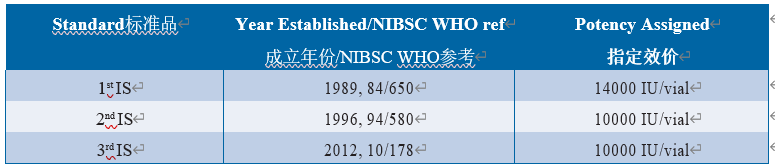
Table 6.5.2-1 Endotoxin standards and their assigned potency内毒素国际标准品及效价(仅供学习交流用)
虽然本技术报告重点关注用于检测胃肠外药物的内毒素标准品,但医疗器械使用大肠埃希菌O55:B5内毒素(先前可从Difco Laboratories获得)作为内毒素标准品。健康工业制造协会(HIMA)提倡采用O55:B5标准。幸运的是,一项主要合作研究表明,当用LAL(114)检测时,O55:B5内毒素制剂与O113:H10:K(-)制剂一样有效。目前,O55:B5制剂通常用作内毒素对照标准品(CSE)。
热原常用方法试验(即RPT)和内毒素检测(即LAL)本质上是生物学方法,它们都具有明显可变性(实验室间和批次间)。因此,长达40年的科学研究得出结论,纯化、有效和充分表征的LPS作为RSE的最佳分析物。由于RSE(一级标准)的可用性有限,因此不计划用于制药行业的日常使用。而是使用CSE(二级标准品)用于药品生产的日常检测。这些CSE必须使用来源有限的RSE进行标定。官方药典通常不提供制定二级标准品或工作标准品的指南,因为官方药典处于法律认可和官方地位需在发生争议时必须使用药典标准进行仲裁(The official compendia typically do not provide guidance for the establishment of secondary or working standards due to the legal recognition and official status associated with these standards that obligate the use of a pharmacopeial standard with a referee procedure in the case of a dispute.)(裁判不好下场做运动员)。鉴于没有指南,CSE应同RS一样具备相同特征;即它们是耐用的、可靠的、稳定的,并且来自确定的菌株。这些特性是近一个世纪的经验积累,包括各种制剂以及对LPS与人类先天免疫系统的结构和生化相互作用的生物和生化研究。由于药物生产中可能的污染源不可能预先知道,因此使用可鉴别LER现象的内毒素参比品至关重要。来自大肠埃希菌的纯化LPS已被鉴定为对LER敏感(30;41)。在本指南发表时已经开始了EC-7的生产,并将使用与EC-6相同来源的原材料【大肠埃希菌O113:H10:K(-)】。
6.6Natural Occurring Endotoxins (NOEs)自然存在的内毒素
新创造的术语“天然内毒素”(NOE)是指在极少的非化学处理(例如离心和过滤)的条件下由革兰氏阴性菌制备的细菌内毒素制剂。以这种方式制备的内毒素旨在反映生产环境中出现的内毒素。Schwarz及其同事还报告,NOE和化学纯化的LPS内毒素标准品均可受到LER的影响(115)。有关LER机制的信息,请参见技术报告第5.0节。
内毒素对掩蔽的敏感性可能部分归因于类脂A的结构,因类脂A驱动了其生物物理状态组装的热力学。如第3.3节所述,各种革兰氏阴性微生物根据其生长和/或环境条件改变其类脂A结构。实例研究3“放置时间研究中纯化和非纯化内毒素的使用”(第8.3节)中描述的研究表明,遮蔽敏感性取决于细菌种类,并且培养条件确实影响内毒素的遮蔽敏感性。此外,这项研究表明,分离方法不会改变脂质A的一级结构(即NOE的脂质A图谱与其高度纯化的对应物相似);但是,内毒素的超分子结构可能受到制备方法的影响。
由于用于生成NOE的方法和微生物可能因实验室而异,使用NOE可能使在细菌内毒素检查过程中发现变异性进一步复杂化。例如,Bowers和Tran使用0.45 μm过滤从各种微生物中生成NOE(116),其他实验室通过使用替代过滤方法或通过高压灭菌或其他热处理生成NOE。任何一个实验室可能选择的各种技术和内毒素使人想起早期LAL检测。例如,Levin及其同事使用购自Difco Laboratories(Detroit,MI)的大肠埃希菌内毒素和弧菌内毒素粗品初步描述了内毒素诱导的鲎试剂和鲎变形细胞溶解物血液凝固,尽管这些研究是在建立内毒素单位(EU)之前进行的(94)。自然生长条件在不同革兰氏阴性细菌种类之间的内毒素异质性中也发挥着重要作用,使用特定的标准品,如RSE或校准的CSE进行细菌内毒素检查消除了检测方法中引入的额外变异。
6.7Immune Response免疫反应
细菌内毒素可产生广泛的显著病理生理效应,涉及患者安全,例如凝血和补体激活以及激肽释放酶/激肽系统级联反应。因此,除了与最近关于增加对内毒素如何激活免疫系统的理解的进展相关的致热原性外,还需要考虑内毒素的生物学效应。
根据晶体结构,脂多糖单体与MD-2结合,然后lps-MD-2复合物与TLR4受体二聚体结合(图6.7-1)(82)。该受体-配体复合物的形成导致信号级联的激活,从而产生促炎性细胞因子。这些趋化因子诱使免疫细胞到先天性免疫系统被激活的位置诱发炎症(117-121)。活化的巨噬细胞和中性粒细胞错误地聚集到炎症部位,以受控方式杀死入侵的病原体并结束感染。然而,该通路的过度激活可导致严重的后果,甚至是致命的。
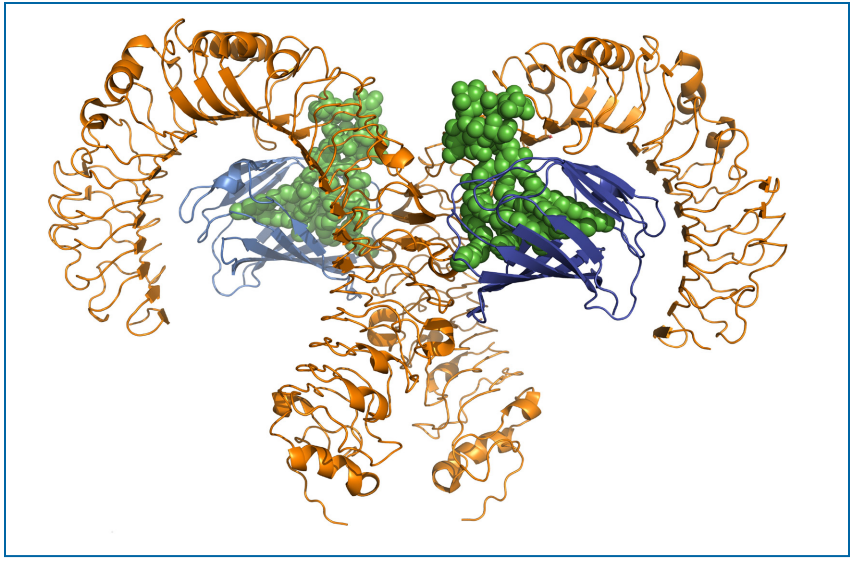
Figure 6.7-1 Structural Basis of Lipopolysaccharide Recognition by TLR4-MD-2 Complex (LPS = green; TLR4 = orange; MD-2 = blue) Figured modified from original.(图片来源于PDA TR82,仅供学习交流用)
少量内毒素可导致免疫反应;因此,确保制造过程最大限度地减少潜在的内毒素和细菌污染是当务之急。有多种病原体相关分子(PAMP),如鞭毛蛋白、孔蛋白和核酸,可诱导免疫应答。在存在病原体或PAMP的情况下,免疫系统使用强大的方法防御感染。PAMPs结合受体,反过来激活先天免疫系统,这对于抵御入侵的病原体至关重要(图6.7-2)(122)。原发性炎症反应可以是局部的,也可以是全身性的,可以提供对病原体的有益防御。内毒素激活TLR4通路除了作为抵抗革兰阴性菌的主要和快速防线的功能外,还可引发获得性免疫。因此,内毒素与人类蛋白MD2和TLR4之间的分子相互作用可以启动先天和适应性系统。
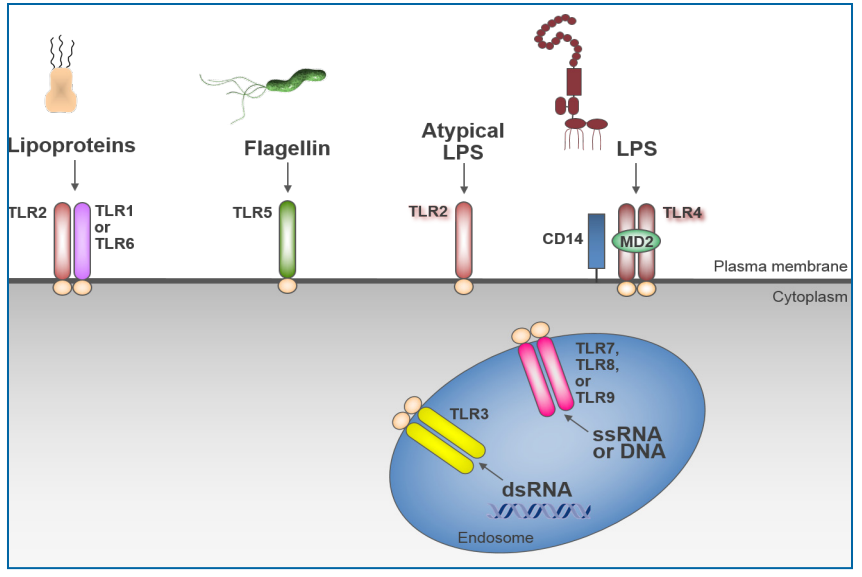
Figure 6.7-2 TLR Recognition of Microbial Components, adapted from Liu, et al.(图片来源于PDA TR82)
当在血液循环中检测到LPS浓度升高时,上皮细胞和免疫细胞中TLR4介导的固有免疫信号级联被激活。活化的细胞反过来产生促炎趋化因子和细胞因子,如IL-1、IL-6、IL-18和TNF-。使用小鼠模型的研究使用了1 mg/kg至25 mg/kg以模拟人或重度感染血液中高浓度内毒素的生理效应(123)。相比之下,人类志愿者静脉给予RSE(2-20 EU/kg)的研究表明,对低水平内毒素暴露的反应并不严重(124-127)。然而在该范围内给药内毒素已被证明可诱导发热、不适、血液中细胞因子水平升高和心脏功能改变(68-72)。
在正常情况下,肠道菌群产生LPS,由于肠道通透性内LPS可能会进入体循环。然而从肠道进入门静脉循环的LPS被肝脏解毒(73-76)。不内毒素肝素抗凝的血浆热处理后测得的循环血液中内毒素水平范围为0-10 pg/mL(128,129)。肝素化热处理“血清”中的内毒素水平范围为0-12.4 pg/mL,EDTA抗凝血浆中报告的内毒素水平范围为0-0.2 ng/mL(130,131)。意外胃肠外给药内毒素可规避这种关键的肝脏保护措施,并可产生多种生理效应。1998年,静脉注射庆大霉素的患者爆发了内毒素样反应。在给药后3小时内,患者出现强烈提示内毒素暴露的症状,即发热、寒战抖动、心率加快和血压下降。随后在美国各地出现了同分异构体,所有同分异构体均与一家生产商有关(132)。试验显示庆大霉素的内毒素呈LAL阳性。尽管庆大霉素中的内毒素浓度低于USP限值,但是超说明书给药导致肠外注射的内毒素高于USP限值5 EU/kg/hr。因为内毒素浓度对于了解这一不良事件至关重要,无法准确测量LER引起的内毒素浓度可能使患者处于重大风险中。
内毒素除了是天然免疫的有效激活剂外,还可刺激适应性免疫(133)。参与获得性免疫的细胞(单核细胞、树突状细胞、B细胞、T细胞)对内毒素的反应方式不同。例如,单核细胞分化为巨噬细胞吞噬病原体,并可作为抗原呈递细胞在内毒素存在的情况下诱导适应性免疫应答(134)。此外,内毒素介导的未成熟树突状细胞TLR4信号通路的诱导导致树突状细胞成熟,在此期间树突状细胞吞噬和过程假定的抗原。树突状细胞从上皮迁移到淋巴组织,并作为抗原呈递细胞,其与T细胞和B细胞的多种亚型连接,对处理的抗原产生适应性免疫应答(135)。此外,TLR4信号通路的激活改变了T细胞和B细胞的功能,并通过在内毒素浓度低至皮克每毫升时增强细胞迁移、细胞附着、克隆扩增和抗体产生来调节对病原体的适应性免疫应答(136,137)。
由于其刺激适应性免疫应答的效力,内毒素可作为疫苗的佐剂;然而,内毒素毒性阻止了其用作活性佐剂。单磷酰脂质A(MPLA)是一种脂质A制剂,来源于明尼苏达沙门氏菌R595 LPS(138)。MPLA具有与LPS相同的基本脂质A结构,并与MD-2/TLR4受体复合物结合。与LPS不同,MPLA缺乏在LPS上发现的几种修饰,效力为(139-141)。尽管效力降低,但MPLA具有免疫刺激作用,在商业应用中用作佐剂,以改善对疫苗的免疫应答(142,143)。
正如MPLA用作疫苗佐剂一样,低浓度内毒素的意外存在可能通过诱导非预期免疫应答而损害胃肠外药物的治疗效果。低剂量的污染物PAMP(如内毒素)可以作为佐剂发挥作用,从而导致对基于蛋白质的药物的意外免疫原性。在一项关于猕猴研究表明,当与治疗蛋白一起转移时,低水平的内毒素(10 pg剂量,平均。重量5.8 kg,1.7 pg/kg)导致产生抗治疗性蛋白的抗体。尽管内毒素剂量不足以诱导全身炎症反应,且低于胃肠外药物的内毒素限度,但其不仅足以激活局部先天性免疫应答,还足以激活获得性免疫(144)。

