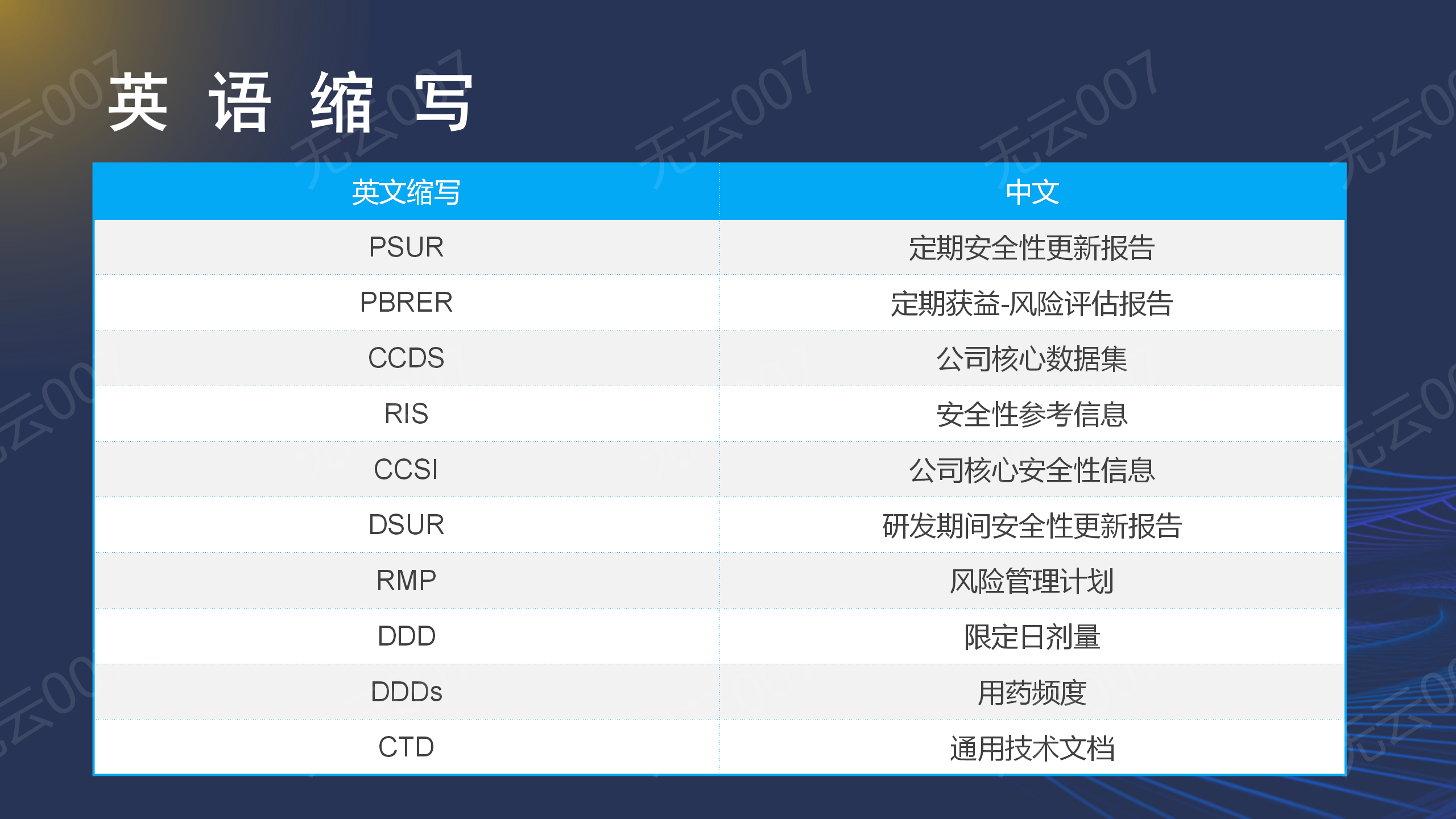
攻略一:英文缩写
学习之前,我们必须要首先学习以下相关的英语缩写,英语缩写的熟悉目前已经成为学习指南以及指导原则的必备基础:,这一块大家自行学习,不过多介绍。
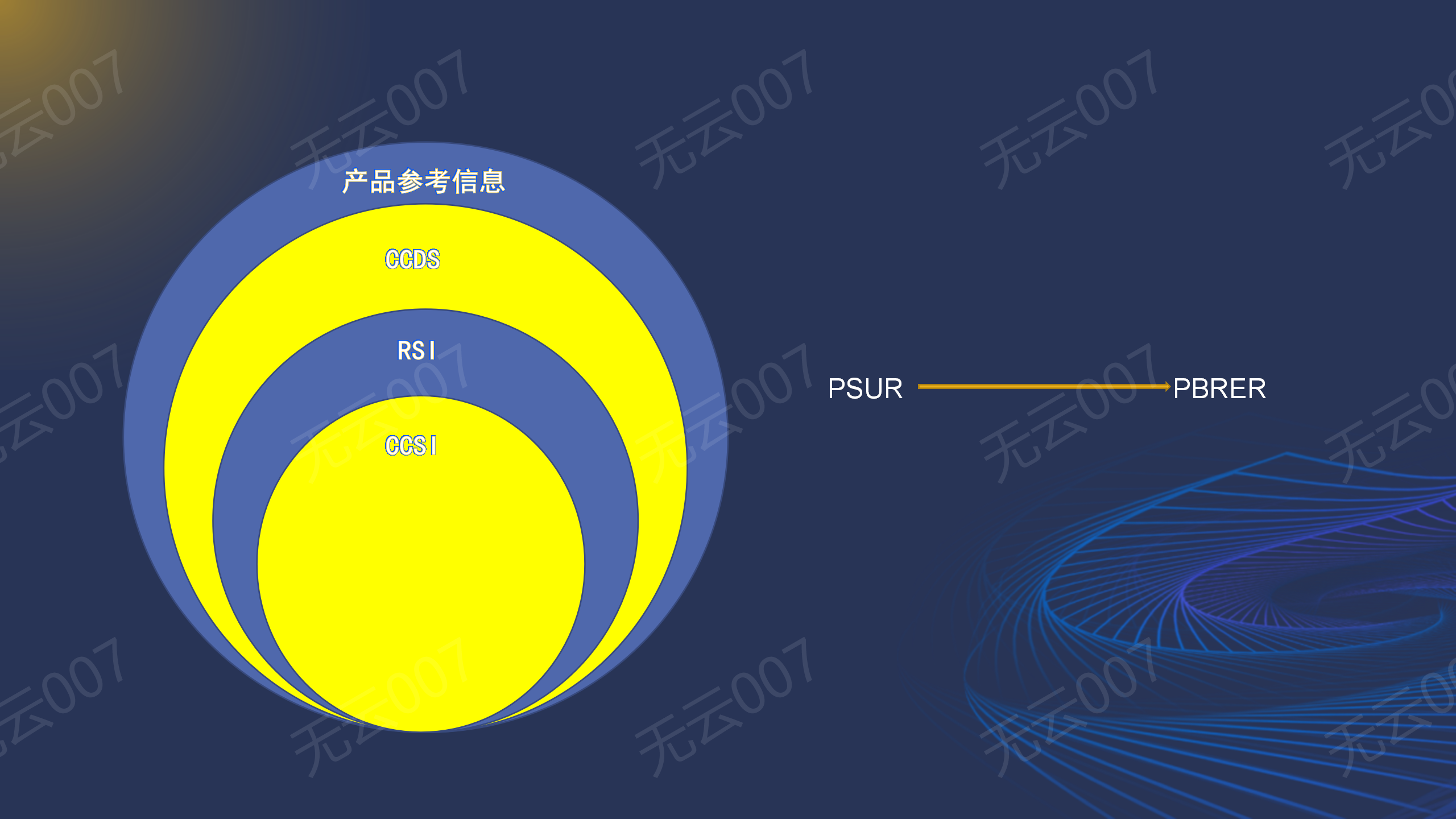
攻略二:关联关系
在这里我们要首先知道PBRER只是PSUR的进阶,PSUR只是报告期内的情况,而PBRER是从始至报告时的所有相关信息,以后随着发展,PBRER是完全有可能替代PSUR的。在撰写这两份报告的同时,均要用接触到产品参考信息、CCDS、RSI、CCSI这之间的关联关系,我个人定义成如下图所示:
攻略三:背景及演变
再说之前,PSUR的背景及演变,还是有必要了解一下:

攻略四:为什么做PSUR/PBRER?
为了解决这个问题,我们首先看需要了解一下,药品上市前后都有哪些特点,看下图:

通过上市后特点我们可以看出PSUR/PBRER的主要工作是解决上市前安全性研究的不足与片面。
攻略五:PSUR的目的
攻略六:《规范》条款再熟悉
《药物警戒质量管理规范》第79条至第86条,这个需要大家私下熟悉。


攻略七:PSUR/PBRER的基本原则与一般要求
1、关于同一活性成分产品的报告;
2、关于复方制剂的报告;
3、关于多家持有人制造或销售的产品;
4、关于报告的数据截止点和提交时间;
5、关于报告的频率;
6、关于报告及附件的提交;
7、关于涉及提交PSUR/PBRER的药品;
8、关于报告的审核
攻略八:安全性参考信息
攻略九:报告格式与内容要求
1、PSUR的格式与内容;




2、PBRER的格式与内容;
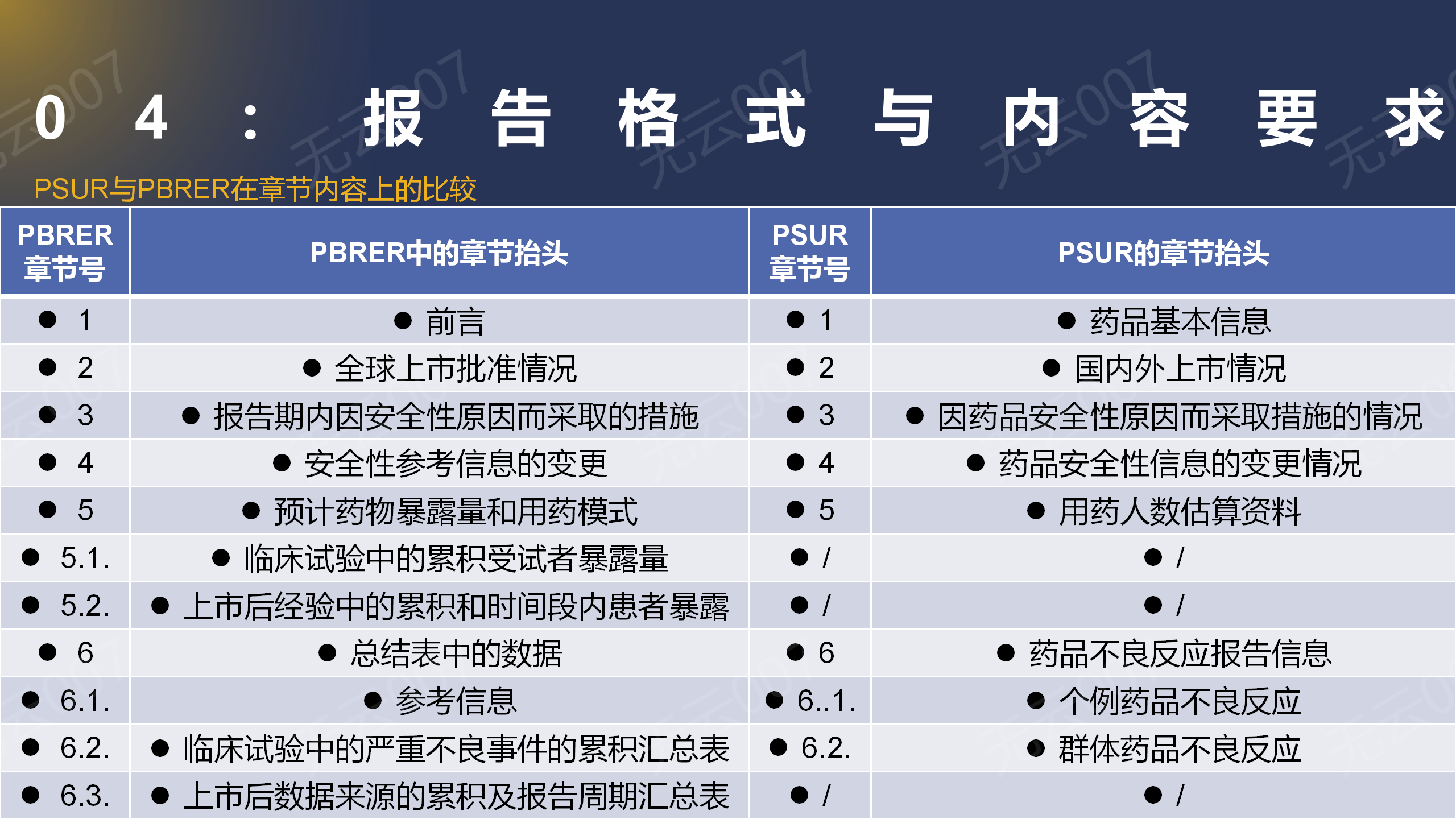
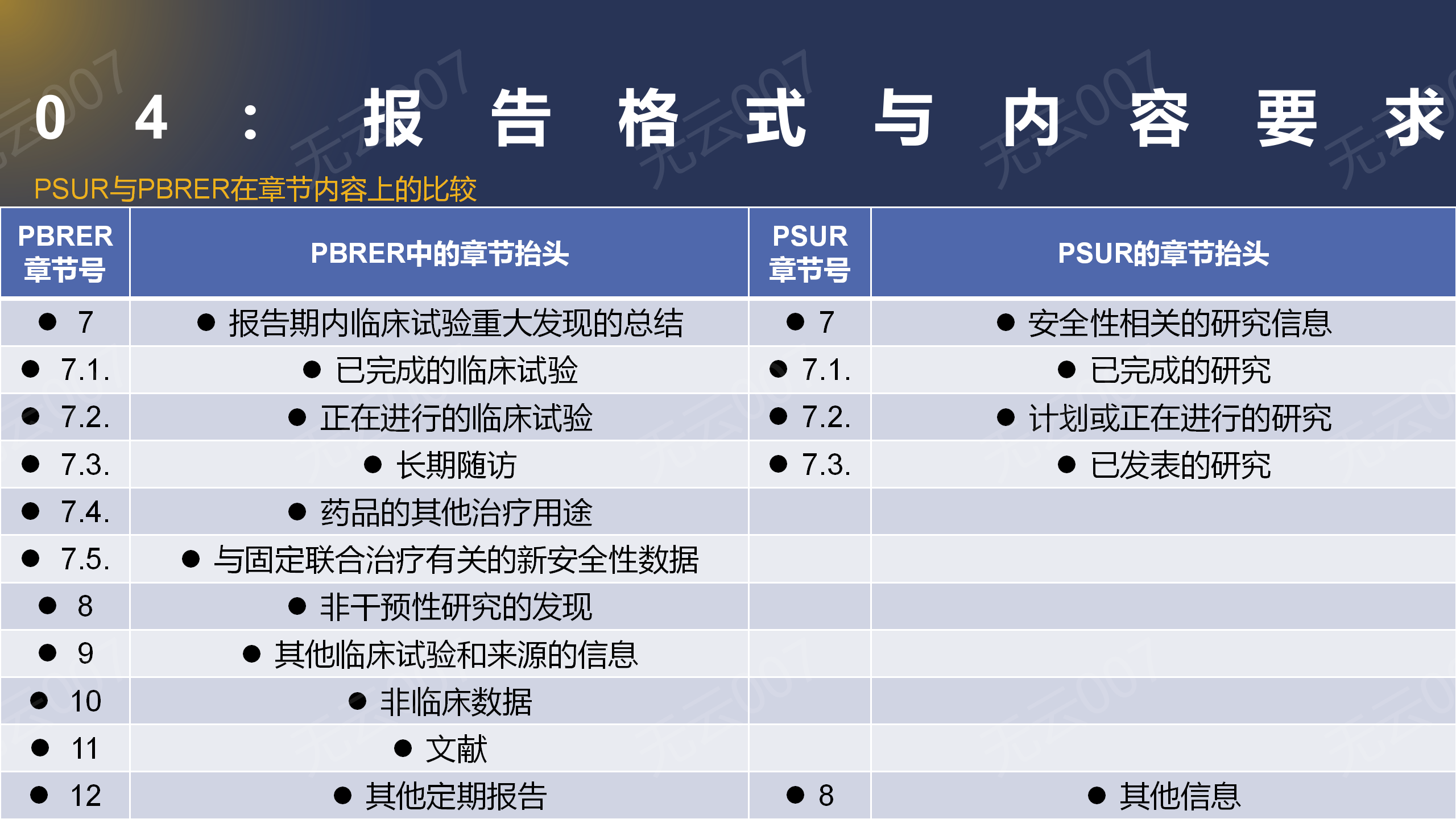
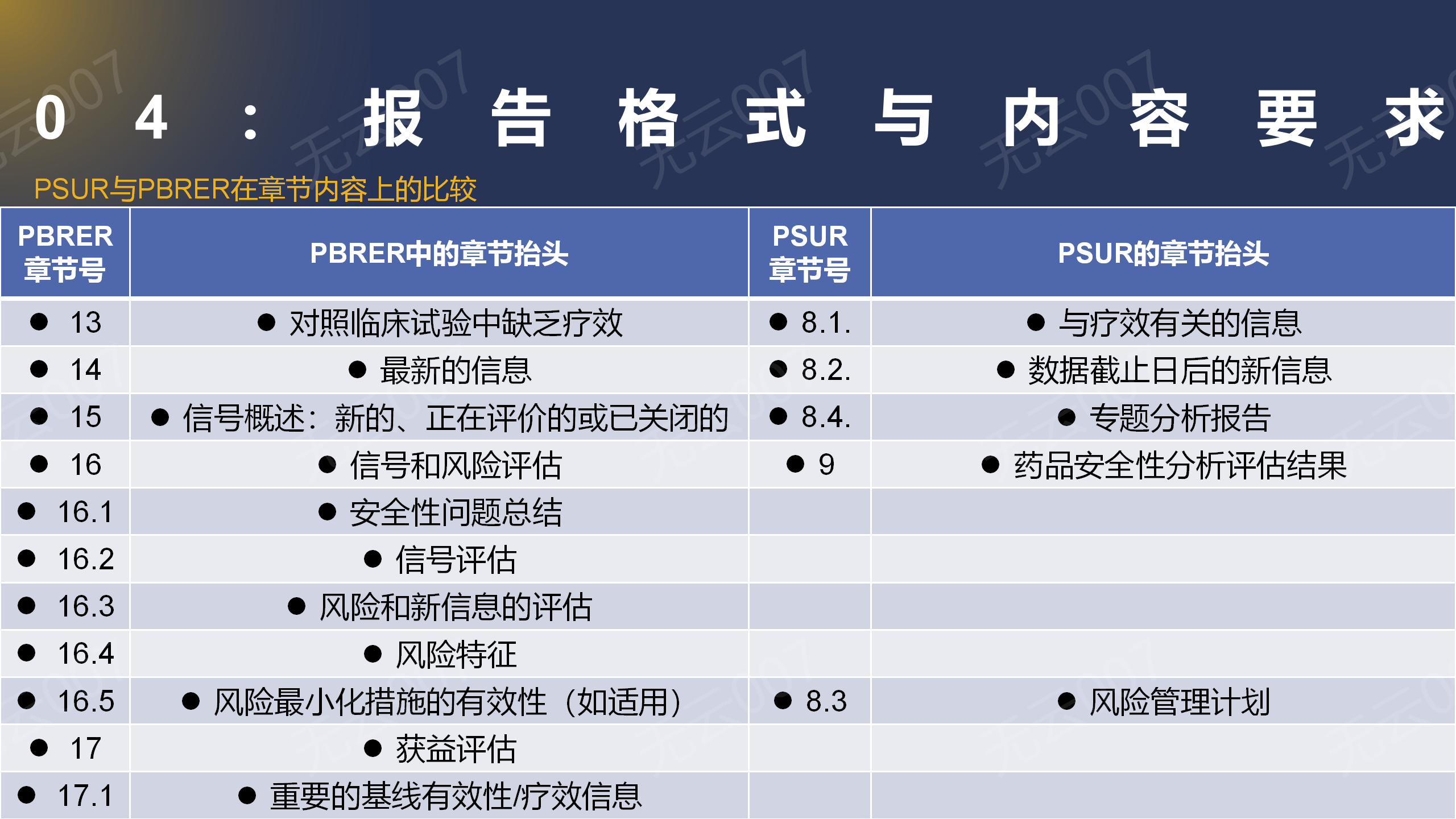
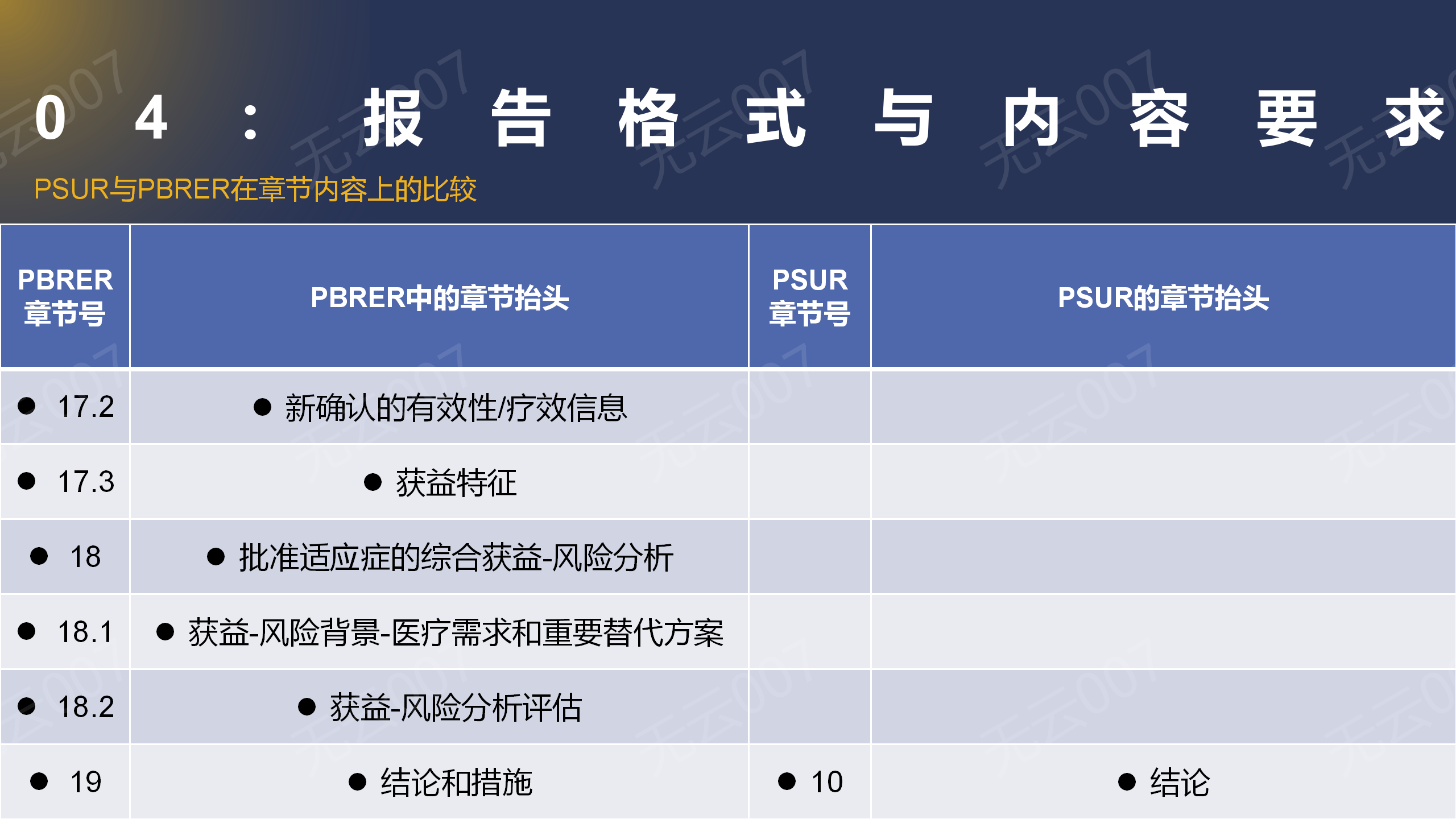
3、PSUR与PBRER在章节内容上的比较