欢迎访问『 博普智库 』制药人必备知识工具



0713 脂肪与脂肪油测定法
 纠错
纠错
本法适用于供药用或药用辅料的脂类物质及类似物(不包括挥发油)的测定。
液体供试品如因析出硬脂发生浑浊时,应先置50℃的水浴上加热,使完全熔化成澄清液体;加热后如仍显浑浊,可离心沉降或用干燥的保温滤器滤过使澄清;将得到的澄清液体搅匀,趁其尚未凝固,用附有滴管的称量瓶或附有玻勺的称量杯,分别称取下述各项检验所需的供试品。固体供试品应先在不高于其熔点10℃的温度下熔化,离心沉降或滤过,再依法称取。
相对密度 照相对密度测定法(通则0601)( (通则0601))测定。
(通则0601))测定。
(通则0601))测定。 折光率 照折光率测定法(通则0622)((通则0622))测定。
(通则0622))测定。 熔点 照熔点测定法(通则0612第二法)((通则0612第二法))测定。
(通则0612第二法))测定。 酸值 酸值系指供试品1g中含有的游离脂肪酸所需氢氧化钾的重量(mg)勘误酸值系指中和供试品1g中含有的游离脂肪酸所需氢氧化钾的重量(mg)。
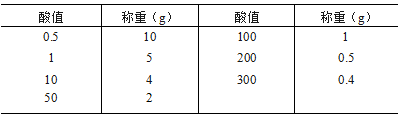
除另有规定外,按表中规定的重量,精密称取供试品,置 250m l锥形瓶中,力口乙醇-乙醚(1 : 1)混合液[临用前加鼢酞指示液1.0ml, 用氢氧化钠滴定液(0. lmol/L )调至微显粉红 色 ] 50ml, 振摇使完全溶解(如不易溶解,缓慢加热回流使溶解),用氢氧化钠滴定液(0. 1mol/L )滴定,至粉红色持续3 0秒不褪。以供试品消耗氢氧化钠滴定液(0. lmol/L )的体积(ml)为 A ,供试品的重量(g)为 W,照下式计算酸值:
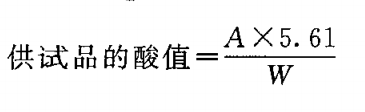
羟值 羟值系指供试品 l g 中含有的羟基,经用以下方法酰化后,所需氢氧化钾的重量(mg)。
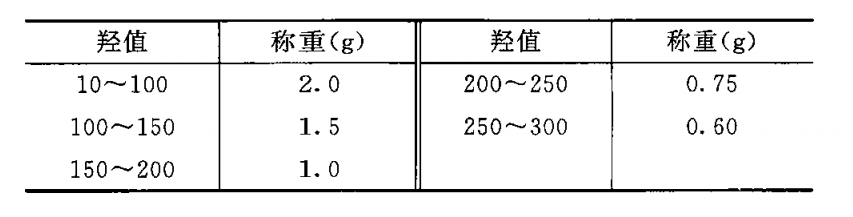
除另有规定外,按表中规定的重量,精密称取供试品,置 250m l的干燥碘瓶中,精密加人酰化剂(取对甲苯磺酸H .4g,置 500m l碘瓶中,加乙酸乙酯360ml,振摇溶解后,缓缓加人醋酐120m l,摇匀,放 置 3 日后用)5ml,用吡啶少许湿润瓶塞,稍拧紧,轻轻摇动使完全溶解,置 50°C 士 1°C水浴中2 5分钟(每 1 0 分钟轻轻摇动)后 ,放冷,加吡啶-水(3 : 5)20ml,5 分钟后加甲酚红-麝香草酚蓝混合指示液 8〜 1 0滴 ,用氢氧化钾(或氢氧化钠)滴定液(lmol/L )滴定至溶液显灰蓝色或蓝色;同时做空白试验。以供试品消耗的氢氧化钾(或氢氧化钠)滴定液(1mol/L )的体积(mol)为 A ,空白试验消耗的体积(ml)为 B ,供试品的重量(g)为 W,供试品的酸值为D ,照下式计算羟值:
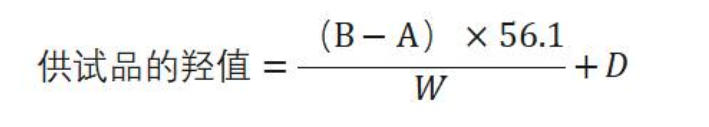
碘值 碘值系指当供试品 100g 充分卤化时所需的碘量(g)。
除另有规定外,取供试品适量 [其重量 (g)约相当于25/供试品的最大碘值],精密称定,置 250m l的干燥碘瓶中,加 三 氯 甲 烷 l 〇ml,溶 解 后 ,精 密加入溴化碘溶液25ml, 密塞,摇匀,在暗处放置30分钟。加人新制的碘化钾试液 10m l与 水 100ml,摇 勻 ,用 硫 代 硫 酸 钠滴定液 (0. 1mol/L )滴定剩佘的碘,滴定时注意充分振摇,待混合液的棕色变为淡黄色,加淀粉指示液lm l,继续滴定至蓝色消失;同时做空白试验。以供试品消耗硫代硫酸钠滴定液 (0. 1mol/L )的体积(ml)为 A ,空白试验消耗的体积(ml)为B ,供试品的重量(g)为 W,照下式计算碘值:
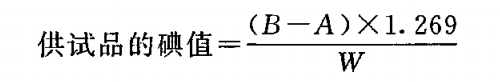
过氧化值 过氧化值系指供试品1000g 中含有的其氧化力与一定量的氧相当的过氧化物量。
除另有规定外,取供试品 5g ,精密称定,置 250ml碘瓶中,加三氯甲烷-冰醋酸(2 : 3)混 合 液 30ml, 振摇溶解后 ,加人碘化钾试液〇.5ml,准确振摇萃取1 分钟,然后加 水 30ml, 用硫代硫酸钠滴定液(0.01mol/L )滴定,滴定时,注意缓慢加人滴定液,并充分振摇直至黄色几乎消失,加淀粉指示液5ml, 继续滴定并充分振摇至蓝色消失,同时做空白试验。空白试验中硫代硫酸钠滴定液(0.01lmol/L )的消耗 量不得过0. 1m l。以供试品消耗硫代硫酸钠滴定液 (0. 01mol/L )的体积(ml)为 A , 空白试验消耗硫代硫酸钠滴定液(0. 01m〇l/L )的体积(ml)为 B ,供试品的重量(g)为 W,
照下式计算过氧化值:
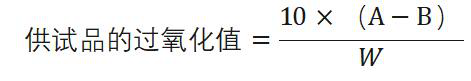
皂化值皂化值系指中和并皂化供试品l g 中含有的游离酸类和酯类所需氢氧化钾的重量(mg)。
除另有规定外,取 供 试 品 适 量 [其重量 (g)约相当于250/供试品的最大皂化值],精密称定,置 250m l回流瓶中,精密加入 0. 5mol/L 氢氧化钾乙醇溶液25ml,加热回流30分钟,然后用乙醇l 〇m l冲洗冷凝器的内壁和塞的下部,加酚酞指示液1. 0ml,用盐酸滴定液(0. 5mol/L )滴定剩余的氢 氧化钾,至溶液的粉红色刚好褪去,加热至沸,如溶液又出 现粉红色,再滴定至粉红色刚好褪去;同时做空白试验。以供试品消耗的盐酸滴定液(〇. 5m〇l/L )的体积(ml)为 A ,空白试验消耗的体积(ml)为 B ,供试品的重量(g)为 W,照下式计算皂化值:
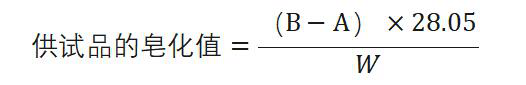
不皂化物 除 另 有 规 定 外 ,取供试品约5g ,精密称定, 置 250m l回流瓶中,加氢氧化钾乙醇溶液(取氢氧化钾12g , 加水10m l溶解,用乙醇稀释至100ml,摇匀)50ml,水浴加热回流1 小时,放 冷 至 25°C 以下,移至带有聚四氟乙烯活塞的分液漏斗中,用水洗涤回流瓶 2 次 ,每 次 50ml,洗液并入分液漏斗中。用乙醚提取3 次 ,每 次 100ml; 合并乙醚提取液,用水洗涤乙醚提取液3 次,每 次 40ml,静置分层, 弃去水层;依 次 用 3 % 氢氧化钾溶液与水洗涤乙醚层各3次 ,每次40ml,再用水40m l反复洗涤乙醚层直至最后洗液 中加酚酞指示液2滴不显红色。转移乙醚提取液至已恒重的蒸发皿中,并用乙醚10m l洗涤分液漏斗,洗液并入蒸发皿 中,置 50°C水浴上蒸去乙醚,用 丙 酮 6m l溶解残渣,空气流下挥去丙酮。在 l 〇5°C 干燥至连续两次称重之差不超过1m g,计算不皂化物。
取干燥后的残渣,用中性乙醇20ml溶解残渣,加酚酞指示液数滴,用乙醇制氢氧化钠滴定液(0.1mol/L)滴定至粉红色持续30秒不褪色,如果消耗乙醇制氢氧化钠滴定液(0.1mol/L)超过0.2ml,残渣总量不能当作不皂化物重量,试验必须重做。
甾醇组成 取不皂化物项下经乙醇制氢氧化钠滴定液(0.1mol/L)滴定至终点且满足要求的溶液,水浴蒸干,残渣加丙酮6ml溶解,室温挥发至干,残渣在105℃干燥约15分钟,作为供试品。另取葵花籽油,同法制备不皂化物并同法处理,作为对照。
甾醇的分离 取供试品,用乙醚溶解3次,每次4ml,转移至试管中,氮气流下挥发至干,加流动相适量溶解残渣(必要时,可加异丙醇1~3滴以促溶),制成每1ml中约含残渣40mg的溶液,用0.45μm滤膜滤过,取续滤液作为供试品溶液;另取上述对照,同法操作,作为对照溶液;取胆固醇和β-谷甾醇各适量,分别加流动相溶解并稀释制成每1ml中约含40mg的溶液,作为胆固醇和β-谷甾醇定位用溶液。照高效液相色谱法(通则0512)((通则0512))试验,用硅胶为填充剂(250mm×4.6mm,5μm;预柱5mm×4.6mm,5μm),以异丙醇-正己烷(1:99)为流动相,流速为每分钟1.0ml,检测波长为210nm。取对照溶液、供试品溶液、胆固醇和β-谷甾醇定位用溶液各50μl,分别注入液相色谱仪,记录色谱图,对照溶液应在23~32分钟显示两个主要的色谱峰,收集对照溶液、供试品溶液、胆固醇和β-谷甾醇定位用溶液约20分钟至32分钟间的洗脱液(注:收集起始时间以胆固醇的出峰时间为准),分别置试管中,每个试管收集两次进样所得的洗脱液,氮气流下挥发至干。
(通则0512))试验,用硅胶为填充剂(250mm×4.6mm,5μm;预柱5mm×4.6mm,5μm),以异丙醇-正己烷(1:99)为流动相,流速为每分钟1.0ml,检测波长为210nm。取对照溶液、供试品溶液、胆固醇和β-谷甾醇定位用溶液各50μl,分别注入液相色谱仪,记录色谱图,对照溶液应在23~32分钟显示两个主要的色谱峰,收集对照溶液、供试品溶液、胆固醇和β-谷甾醇定位用溶液约20分钟至32分钟间的洗脱液(注:收集起始时间以胆固醇的出峰时间为准),分别置试管中,每个试管收集两次进样所得的洗脱液,氮气流下挥发至干。甾醇的测定 避免潮湿 。取留醇的分离项下供试品溶液制得残渣,加无水吡啶0.2ml,加 N,〇-双 (三甲基硅烷)三氟乙酰胺(BSTFA)-三甲基氯硅烷(TM CS)(99 : 1)混合液0.2ml,密封,混匀,80°C加 热 2 0分钟,取出,放冷,取液 体层作为供试品衍生化溶液。另取阁醇的分离项下对照溶液 、胆固醇与P-谷留醇定位用溶液制得残渣,分别自 “加无水吡啶 0.2ml”起同法操作,取液体层分别作为对照衍生化溶液、胆固醇衍生化溶液和卩-谷留醇衍生化溶液。照气相色谱法(通则 0521)((通则 0521))测定,采 用 以 5 % 苯基- 9 5 %甲基聚硅氧烷为固定液的毛细管色谱柱(30m X 0. 25mm, 0. 25pm),以氦气为载气,起始温度为 260°C , 维持 5 0分钟,以每分钟5°C的速率升温至290°C , 维 持 5 分钟,进样口温度为 290°C ,检测器温度为290°C 。取对照衍生化溶液1〜 ( 视甾醇量而选择),注人气相色谱仪,记录的色谱图中,应显示4个主要的色谱峰,分别为菜油甾醇峰、豆甾醇峰、 谷甾醇峰和 A7-豆甾醇峰,菜油留醇峰与豆甾醇峰的分离度应不小于4.0。另取与对照衍生化溶液相同进样体积的胆固醇衍生化溶液、β谷留醇衍生化溶液和供试品衍生化溶液,分别注入气相色谱仪,记录色谱图,按下表所附的相对β谷留醇峰的保留时间鉴别各甾醇峰,计 算 从 胆 固 醇 到 △7-燕麦甾醇 15个峰的总峰面积,按峰面积归一化法计算供试品中各留醇的含量。
(通则 0521))测定,采 用 以 5 % 苯基- 9 5 %甲基聚硅氧烷为固定液的毛细管色谱柱(30m X 0. 25mm, 0. 25pm),以氦气为载气,起始温度为 260°C , 维持 5 0分钟,以每分钟5°C的速率升温至290°C , 维 持 5 分钟,进样口温度为 290°C ,检测器温度为290°C 。取对照衍生化溶液1〜 ( 视甾醇量而选择),注人气相色谱仪,记录的色谱图中,应显示4个主要的色谱峰,分别为菜油甾醇峰、豆甾醇峰、 谷甾醇峰和 A7-豆甾醇峰,菜油留醇峰与豆甾醇峰的分离度应不小于4.0。另取与对照衍生化溶液相同进样体积的胆固醇衍生化溶液、β谷留醇衍生化溶液和供试品衍生化溶液,分别注入气相色谱仪,记录色谱图,按下表所附的相对β谷留醇峰的保留时间鉴别各甾醇峰,计 算 从 胆 固 醇 到 △7-燕麦甾醇 15个峰的总峰面积,按峰面积归一化法计算供试品中各留醇的含量。 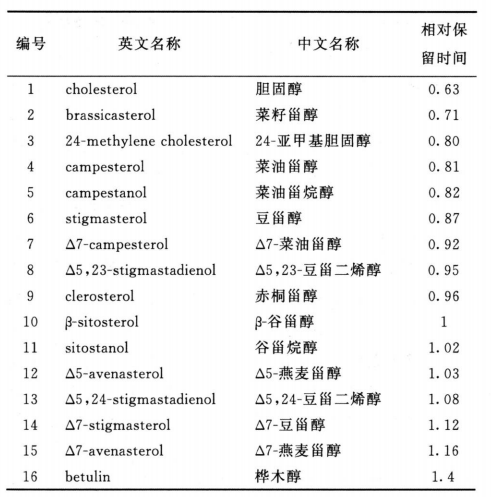
脂肪酸凝点 (1)脂肪酸的提取,取 20% ( g/g)氢氧化钾的甘油溶液 75g,置 800m l烧杯中,加 供 试 品 50g,于150°C 在不断搅拌下皂化1 5分钟 ,放冷至约100°C ,加入新沸的水 500ml, 搅 勻 ,缓缓加入硫酸溶液(1- 4)50ml, 加热至脂肪酸明显分离为一个透明层;趁热将脂肪酸移入另一烧杯中,用新煮沸的水反复洗涤,至洗液加入甲基橙指示液显黄色,趁热将澄清的脂肪酸放入干燥的小烧杯中,加无水乙醇5ml,搅 匀 ,用小火加热至无小气泡逸出, 即得。
(2)凝点的测定 取按上法制得的干燥脂肪酸,照凝点测定法(通则0613)((通则0613))测定。
(通则0613))测定。 脂肪酸组成 除另有规定外,取供试品0.1g,置50ml回流瓶中,加0.5mol/L氢氧化钠甲醇溶液4ml,在水浴中加热回流直至油滴消失(通常约10分钟),放冷,加14%三氟化硼甲醇溶液5ml,再在水浴中加热回流2分钟,放冷,加正庚烷4ml,继续在水浴中加热回流1分钟后,放冷,加饱和氯化钠溶液10ml,摇匀,静置使分层,取上层液,经无水硫酸钠干燥,作为供试品溶液;分别取硬脂酸甲酯、棕榈酸甲酯和油酸甲酯适量,用正庚烷溶解并稀释制成每1ml中各约含0.1mg的溶液,作为系统适用性溶液。照气相色谱法(通则0521)((通则0521))试验,采用以聚乙二醇(或极性相近)为固定液的毛细管色谱柱(30m×0.53mm,1.0μm),起始温度为70℃,维持2分钟,以每分钟5℃的速率升温至240℃,维持24分钟;进样口温度为220℃;检测器温度为260℃。取系统适用性溶液1μl注入气相色谱仪,记录色谱图,棕榈酸甲酯峰和硬脂酸甲酯峰相对于油酸甲酯峰的保留时间分别约为0.87和0.99,理论板数按油酸甲酯峰计算不低于10000,各色谱峰的分离度应符合要求。取供试品溶液1μl,注入气相色谱仪,记录色谱图,按峰面积归一化法计算各脂肪酸甲酯的含量。
(通则0521))试验,采用以聚乙二醇(或极性相近)为固定液的毛细管色谱柱(30m×0.53mm,1.0μm),起始温度为70℃,维持2分钟,以每分钟5℃的速率升温至240℃,维持24分钟;进样口温度为220℃;检测器温度为260℃。取系统适用性溶液1μl注入气相色谱仪,记录色谱图,棕榈酸甲酯峰和硬脂酸甲酯峰相对于油酸甲酯峰的保留时间分别约为0.87和0.99,理论板数按油酸甲酯峰计算不低于10000,各色谱峰的分离度应符合要求。取供试品溶液1μl,注入气相色谱仪,记录色谱图,按峰面积归一化法计算各脂肪酸甲酯的含量。 加热试验 取供试品约50ml,置烧杯中,在砂浴上加热至280℃,升温速率为每分钟上升10℃,观察油的颜色和其他性状的变化。
杂质 取供试品约20g,精密称定,置锥形瓶中,加石油醚(沸程60~90℃)20ml使溶解,用干燥至恒重的垂熔玻璃坩埚滤过(如溶液不易滤过,可添加石油醚适量),用石油醚洗净残渣和滤器,在105℃干燥至恒重;精密称定,增加的重量即为供试品中杂质的重量。
水分与挥发物 取供试品约5g,置干燥至恒重的扁形称量瓶中,精密称定,在105℃干燥40分钟取出,置干燥器内放冷,精密称定重量;再在105℃干燥20分钟,放冷,精密称定重量,至连续两次干燥后称重的差异不超过0.001g,如遇重量增加的情况,则以增重前的一次重量为恒重。减失的重量,即为供试品中含有水分与挥发物的重量。
碱性杂质 取新蒸馏的丙酮10ml、水0.3ml和0.04%溴酚蓝乙醇溶液1滴,用0.01mol/L盐酸溶液或0.01mol/L氢氧化钠溶液调节至中性,精密加供试品10ml,摇匀,静置,用盐酸滴定液(0.01mol/L)滴定至上层液显黄色,计算消耗的盐酸滴定液(0.01mol/L)体积。
甲氧基苯胺值 避光快速操作。除另有规定外,取供试品0.5g,精密称定(W),置25ml量瓶中,加异辛烷溶解并稀释至刻度,作为供试品溶液,照紫外-可见分光光度法(通则0401)((通则0401)),以异辛烷为空白,在350nm的波长处测定吸光度(A1);另取10ml具塞试管2支,供试品管加供试品溶液5.0ml,空白管加异辛烷5.0ml,再各加0.25%的4-甲氧基苯胺的冰醋酸溶液1.0ml,振摇,暗处放置10分钟,以空白管溶液作为空白,在350nm的波长处测定供试品管溶液的吸光度(A2)。照下式计算甲氧基苯胺值:
(通则0401)),以异辛烷为空白,在350nm的波长处测定吸光度(A1);另取10ml具塞试管2支,供试品管加供试品溶液5.0ml,空白管加异辛烷5.0ml,再各加0.25%的4-甲氧基苯胺的冰醋酸溶液1.0ml,振摇,暗处放置10分钟,以空白管溶液作为空白,在350nm的波长处测定供试品管溶液的吸光度(A2)。照下式计算甲氧基苯胺值: 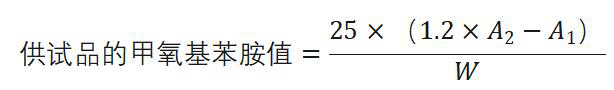
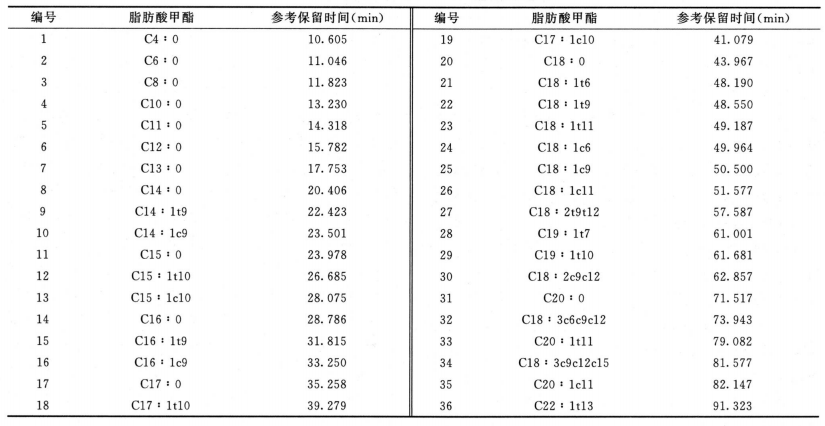
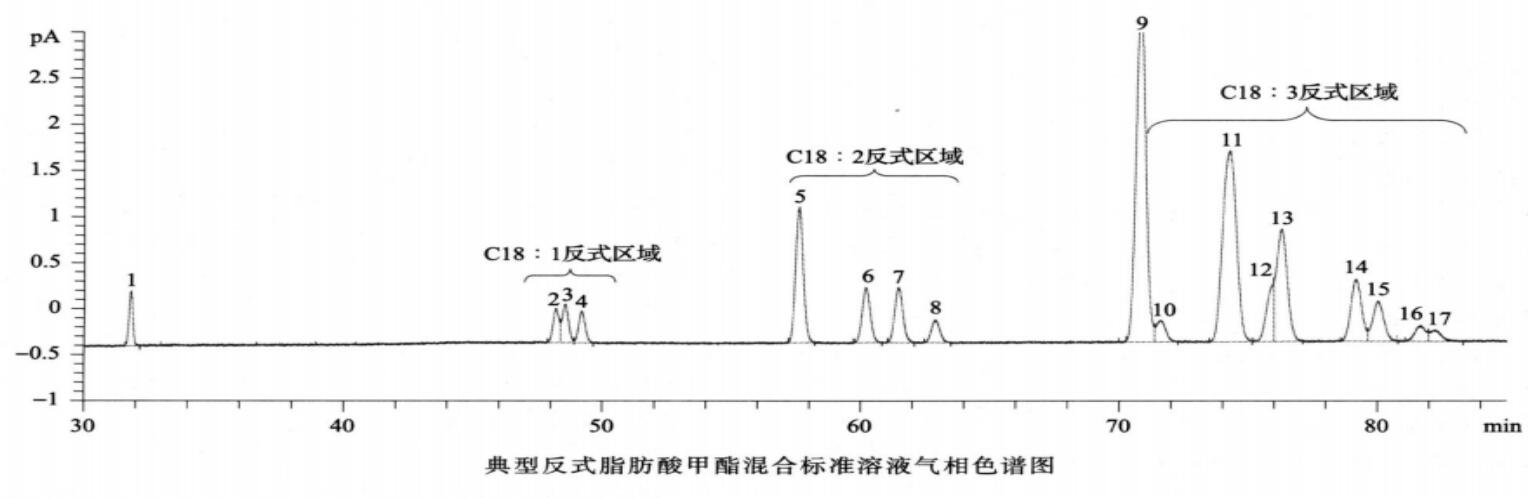
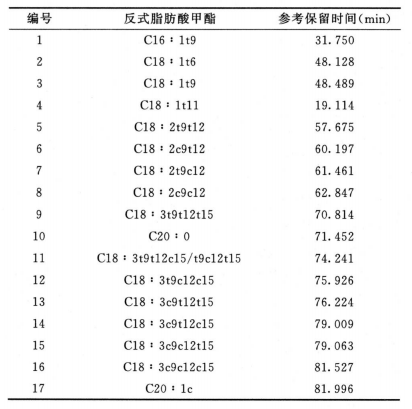
反式脂肪酸 除另有规定外,取供试品100mg,置50ml回流瓶中,加0.5mol/L氢氧化钠甲醇溶液4ml,在水浴中加热回流直至油滴消失(通常约10分钟),放冷,加14%三氟化硼甲醇溶液5ml,再在水浴中加热回流5分钟,放冷,加异辛烷2ml,继续在水浴中加热回流1分钟,放冷,加饱和氯化钠溶液10ml,摇匀,静置使分层,取上层液,经无水硫酸钠干燥,作为供试品溶液。分别取油酸甲酯、反式油酸甲酯、亚油酸甲酯顺反异构体混合溶液和亚麻酸甲酯顺反异构体混合溶液适量,加异辛烷溶解并稀释制成每lml中约含油酸甲酯1mg、反式油酸甲酯1mg、亚油酸甲酯顺反异构体2.5mg、亚麻酸甲酯顺反异构体2.5mg的溶液,作为系统适用性溶液(脂肪酸甲酯分类信息和反式脂肪酸甲酯的参考保留时间见下表)。照气相色谱法(通则0521)((通则0521))试验,釆用以聚二氰丙基硅氧烷(或极性相近)为固定液的毛细管色谱柱(100m×0.25mm,0.2μm),起始温度为163℃,维持85分钟,以每分钟30℃的速率升温至240℃,维持13分钟;分流比45∶1;载气流速:恒压40psi;进样口温度为250℃;检测器温度为250℃。取系统适用性溶液1μl注入气相色谱仪,记录色谱图,顺-9,12-反-15-十八碳三烯酸甲酯(C18 : 3c9c12t15)和亚麻酸甲酯(C18 : 3c9c12cl5)的分离度应不小于1.0(必要时可适当调整色谱系统参数满足上述系统适用性要求,并确保供试品中相应顺反脂肪酸甲酯峰的分离度均不小于1.0;36种脂肪酸甲酯混合标准溶液和典型反式脂肪酸甲酯混合标准溶液的气相色谱图见下图)。取供试品溶液1μl注入气相色谱仪,记录色谱图,按峰面积归一化法计算供试品中各反式脂肪酸甲酯峰占所有脂肪酸甲酯总峰面积的百分含量。
(通则0521))试验,釆用以聚二氰丙基硅氧烷(或极性相近)为固定液的毛细管色谱柱(100m×0.25mm,0.2μm),起始温度为163℃,维持85分钟,以每分钟30℃的速率升温至240℃,维持13分钟;分流比45∶1;载气流速:恒压40psi;进样口温度为250℃;检测器温度为250℃。取系统适用性溶液1μl注入气相色谱仪,记录色谱图,顺-9,12-反-15-十八碳三烯酸甲酯(C18 : 3c9c12t15)和亚麻酸甲酯(C18 : 3c9c12cl5)的分离度应不小于1.0(必要时可适当调整色谱系统参数满足上述系统适用性要求,并确保供试品中相应顺反脂肪酸甲酯峰的分离度均不小于1.0;36种脂肪酸甲酯混合标准溶液和典型反式脂肪酸甲酯混合标准溶液的气相色谱图见下图)。取供试品溶液1μl注入气相色谱仪,记录色谱图,按峰面积归一化法计算供试品中各反式脂肪酸甲酯峰占所有脂肪酸甲酯总峰面积的百分含量。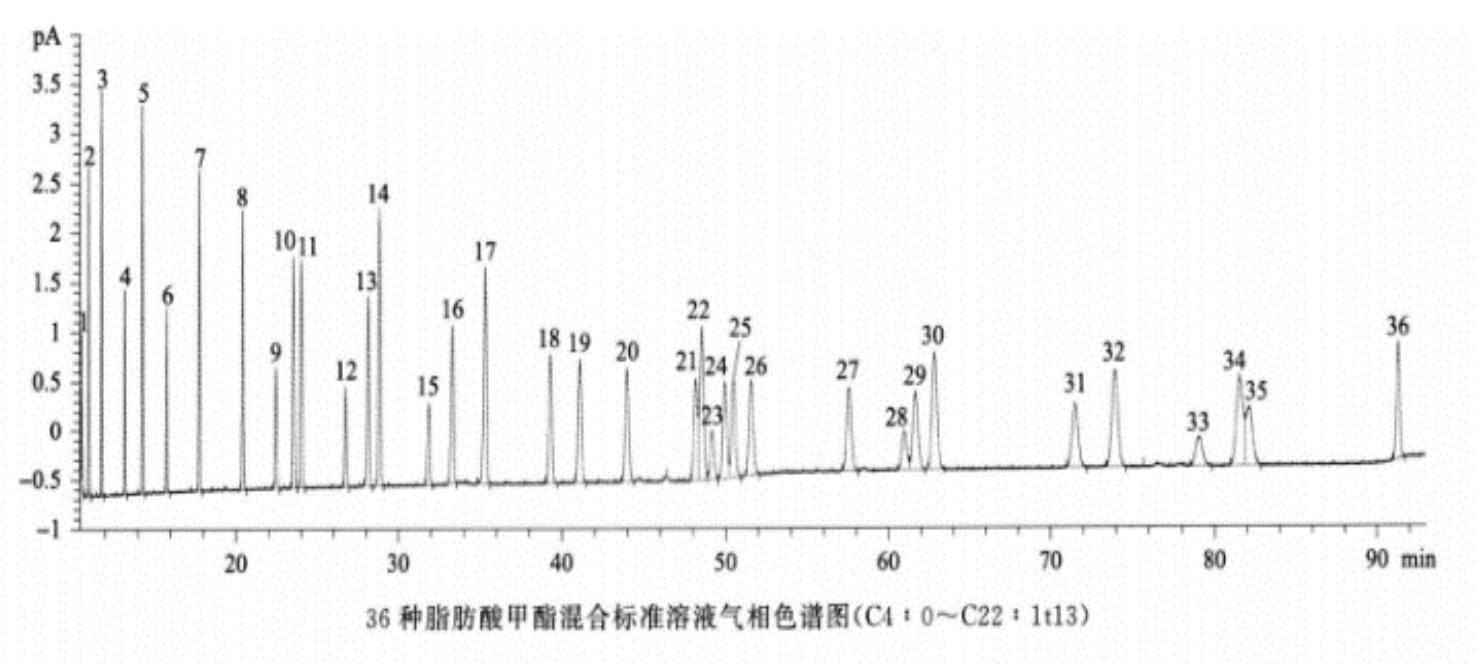
【附注】
1.溴化碘溶液 取研细的碘13.0g,置干燥的具塞玻瓶中,加冰醋酸1000ml,微温使碘完全溶解;另用吸管插入法量取溴2.5ml(或在通风橱中称取7.8g),加入上述碘溶液中,摇匀,即得。为了确定加溴量是否合适,可在加溴前精密取出20ml,用硫代硫酸钠滴定液(0.1mol/L)滴定,记录消耗的体积(ml);加溴后,摇匀,再精密取出20ml,加新制的碘化钾试液10ml,再用硫代硫酸钠滴定液(0.1mol/L)滴定,消耗的体积(ml)应略小于加溴前的2倍。
本液应置具塞玻瓶内,密塞,在暗处保存。
2.乙醇制氢氧化钠滴定液(0.1mol/L) 取50%氢氧化钠溶液2ml,加乙醇250ml,摇匀,即得(如溶液浑浊,配制后放置过夜,取上清液)。取在五氧化二磷干燥器中减压干燥至恒重的基准苯甲酸约0.2g,精密称定,加乙醇10ml与水2ml溶解,加酚酞指示液2滴,用上述滴定液滴定至溶液显持续浅粉红色。每1ml乙醇制氢氧化钠滴定液(0.1mol/L)相当于12.21mg的苯甲酸。
本液应置具橡皮塞的棕色玻瓶中,密闭保存,临用前应标定浓度。
继续阅读
在线查询结果来源于2020年版中国药典,仅供参考。由专业团队进行审核校对。


